ゆるくはあるが軽くはない
S–O 相互作用は、酸素のローンペアが硫黄の反結合性軌道 σ* に供与されることで生じる相互作用で、強さとしては水素結合や π–カチオン相互作用よりは弱いものの、π–π スタッキングや疎水性相互作用よりは強い。硫黄はチオエーテルのような電子豊富な状態よりも、チアゾールのように電子欠乏した環境の方が σ-hole が強くなり、この相互作用を形成しやすい。
BMS 社や Gilead 社は標的タンパク質のアミノ酸主鎖カルボニル基との相互作用に、Accent 社は化合物の分子内で活性配座を固定するために、それぞれこの相互作用を利用して活性を向上させた。
他にも環状ペプチドや PROTAC のカメレオニシティに利用できるかもしれない。
この「ゆるさ」・・・
ゆるくはあるが、軽くはないのだ

【① イントロ: S-O相互作用とは?】
気ままに創薬化学の16年前の記事から引用。
http://www.medchem4410.seesaa.net/article/142718530.html
Intramolecular Nonbonded S···O Interaction Recognized in (Acylimino)thiadiazoline Derivatives as Angiotensin II Receptor Antagonists and Related Compounds.
J. Am. Chem. Soc., 1998, 120, 3104–3110.
https://doi.org/10.1021/ja973109o.
●徳島大学 長尾善光先生らの報告
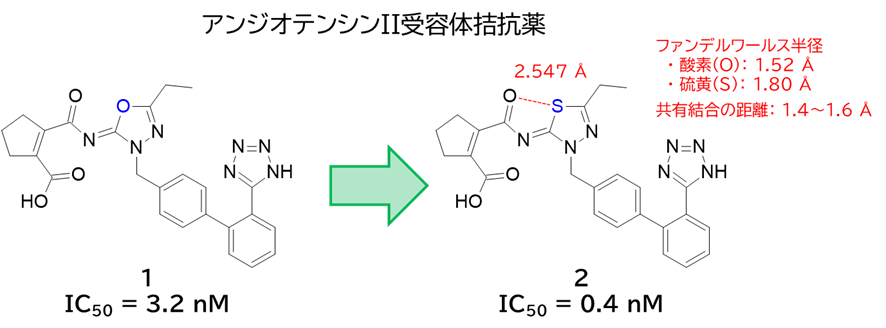
・ 化合物1の(アシリミノ)オキサジアゾリン部分の酸素原子を硫黄原子に置き換えた化合物2は活性が10倍弱も向上
・ (アシリミノ)チアジアゾリン部分にS-O相互作用(約2.5 Å)あり
✓ 硫黄と酸素のファンデルワールス半径の和(3.32 Å)より短い
✓ 共有結合の距離(1.4~1.5 Å)より長い
→ S-O間の相互作用形成が示唆される
Impacts of noncovalent interactions involving sulfur atoms on protein stability, structure, folding, and bioactivity
Org. Biomol. Chem., 2023, 21, 11-23.
https://doi.org/10.1039/D2OB01602H.
●カルフォルニア大学 ヴォルガらのレビュー
・ S-O相互作用は、静電相互作用と分散相互作用の両方が寄与
✓ 静電安定化は、部分的に正に帯電した硫黄(硫黄は電気陰性度が低い)と部分的に負に帯電した酸素との引力によって生じる
✓ ドナー–アクセプター軌道相互作用 (酸素のローンペアが硫黄の反結合性軌道σ*に供与されることで生じる)
・ S-O相互作用の大きさは2~3 kcal/mol
✓ 水素結合(1~10 kcal/mol)やπ–カチオン相互作用(3~8 kcal/mol)より弱い
✓ π-πスタッキング(0.5~2 kcal/mol)や疎水性相互作用(0.5~1 kcal/mol)より強い
ゆるくはあるが軽くはない相互作用と考えられる。
以下、製薬3社の利用事例を見ていく。
【②BMS社のGalectin-3阻害剤でのS-O相互作用】
Identification of benzothiazole derived monosaccharides as potent, selective, and orally bioavailable inhibitors of human and mouse galectin-3; a rare example of using a S···O binding interaction for drug design
Bioorg. Med. Chem., 2024, 101, 117638.
https://doi.org/10.1016/j.bmc.2024.117638
●Galectin‑3(Gal‑3)は、特徴的なCarbohydrate Recognition Domain (CRD)を介してβ‑ガラクトシドと結合するタンパク質で、線維化、がん、炎症など多様な疾患に関与する重要な治療標的である。しかし、Gal‑3 の結合ポケットは浅く親水性で、さらにヒトとマウスで重要残基が異なるため、両方に強力かつ薬物動態的に優れた阻害剤の創出は難しい。既存のTD139 は高い阻害活性を持つが経口吸収性が低く、GB1211は経口吸収性を有するが阻害活性が弱い。
そこでBMS社は、高活性かつ経口吸収性のある化合物の取得を目指した。

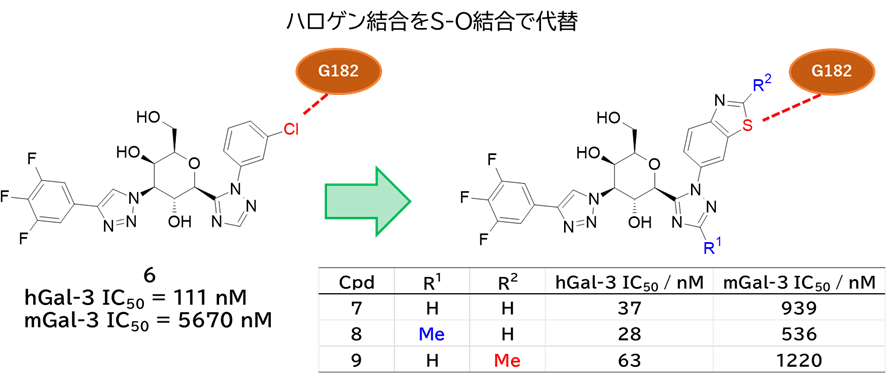
●BMS社が以前に創製した化合物6から展開
・ 化合物6のクロロベンゼンの塩素原子がhGal-3 G182主鎖カルボニル基と相互作用
✓ 硫黄原子に置き換えてS-O相互作用で代替できないか?
→ ベンゾチアゾールに変換した化合物7はhuman/mouseそれぞれ阻害活性が4,5倍向上
・ 周辺展開で、トリアゾールにメチル基の導入が活性向上に有効
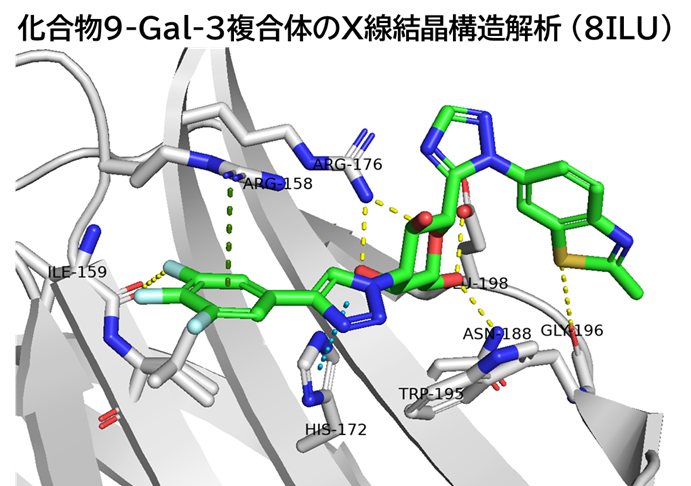
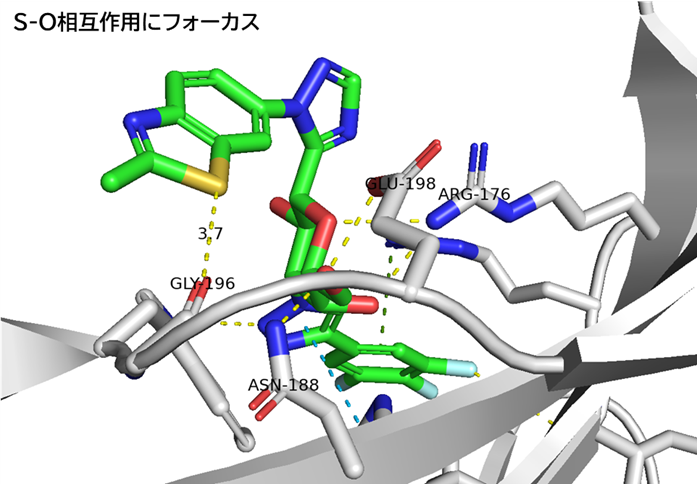
●化合物9-Gal-3複合体のX線結晶構造解析
・ C6ヒドロキシ基とAsn188, Glu198残基と水素結合を形成
・ C5ヒドロキシ基とテトラヒドロピラン酸素原子がArg176と水素結合を形成
・ トリアゾール窒素原子とTrp195残基が水を介して水素結合を形成
・ トリアゾール環とHis172残基がT字型(edge‑to‑face)CH-π相互作用を形成
・ トリフルオロベンゼン環とArg158残基がカチオン-π相互作用を形成
・ テトラヒドロピラン骨格がTrp181残基とファンデルワールス半径相互作用を形成
・ トリフルオロベンゼンC–FとIle159主鎖カルボニル基がorthogonal multipolar interactionを形成
✓ C–F の双極子がカルボニルの双極子に対して直交に配置されると静電的に安定化するらしい
・ C3ヒドロキシ基は相互作用に必須でないかも
→ メチル化して膜透過性を改善できるかも?(脂溶性付与、水素結合ドナー削減)
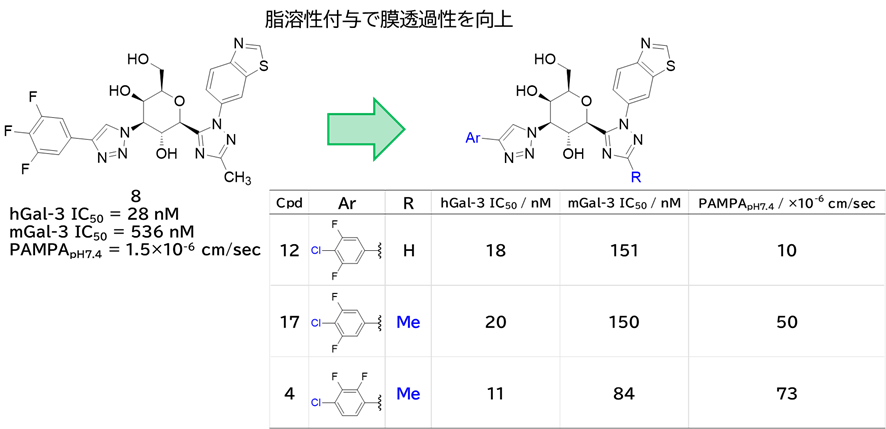
●化合物8に脂溶性付与して膜透過性を上げる
・ まずはトリフルオロベンゼンのp-FをClに変換した化合物12は膜透過性向上
✓ 活性向上は脂溶性効果およびIle159, Val160主鎖カルボニル基とハロゲン結合形成かも
・ C3ヒドロキシ基をメチル化した化合物17は膜透過性向上
✓ 上記構造解析からの狙い通り活性は維持された
・ 芳香環の周辺展開活性によって活性と膜透過性が改善した化合物4を取得
●異なるケミカルクラスの探索
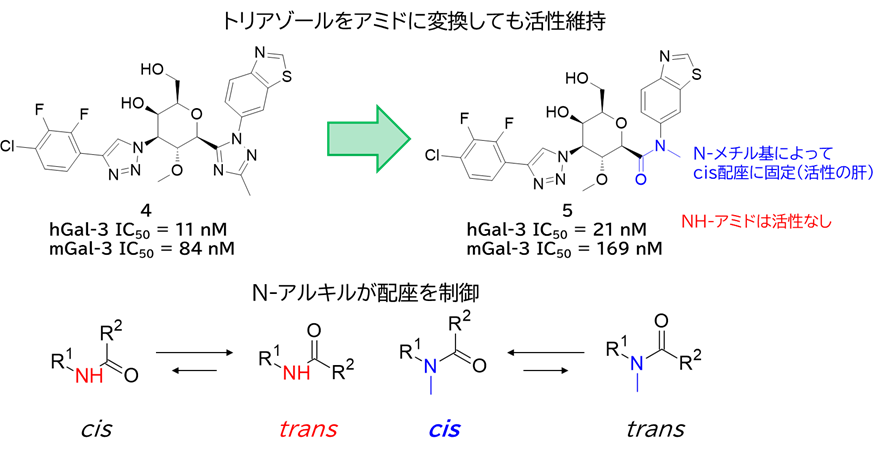
・ 化合物4のC2トリアゾールをアミドに変換
✓ N-メチルアミドで化合物4と同等の阻害活性を有する化合物5を取得
✓ NH-アミドは活性なし、N-エチルは活性あり
→ N-アルキルが配座を制御&トリアゾールのメチルの代替も
→ cis-アミドが活性に必須と考えられる
・ 化合物4,5は共にサブタイプ選択性あり&良好なin vivo PKプロファイル
・ 残念ながらin vivo薬効評価の記載はなし
【③Gilead社のIRAK4阻害剤でのS-O相互作用】
Examination of Noncanonical Kinase Hinge Binders Leads to Thiadiazoles as Potent IRAK4 Inhibitors
ACS Med. Chem. Lett., 2026, 17, 175–182.
https://doi.org/10.1021/acsmedchemlett.5c00602.
●IRAK4(Interleukin‑1 receptor–associated kinase 4)は、IL‑1R/TLR シグナル伝達の中心的キナーゼで、炎症性疾患の治療標的として注目されている。一般的なキナーゼ阻害剤は、キナーゼのヒンジ領域に結合するATPアデニン構造を模倣した典型的 (canonical)なヒンジバインダーによるATP競合的な (オルソステリックな)阻害剤であるが、キナーゼ間の選択性の低さによる副作用や、平面的な構造による物性への影響が懸念される。
そこでGilead社は、アデニン模倣ではないnon-canonicalなヒンジバインダーを探索したところ、ニコチンアミド構造をもつ化合物1を取得した。
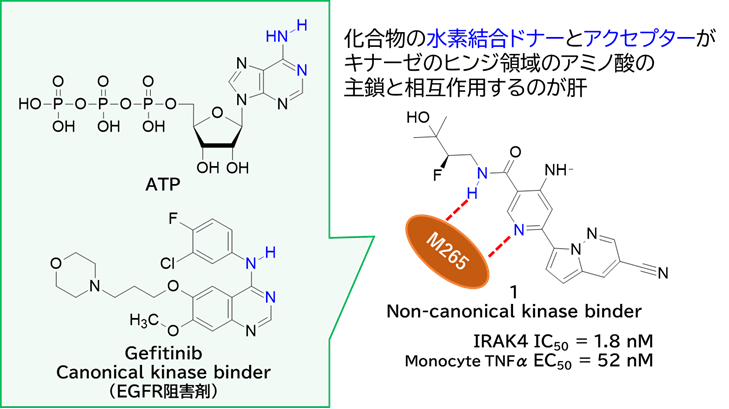
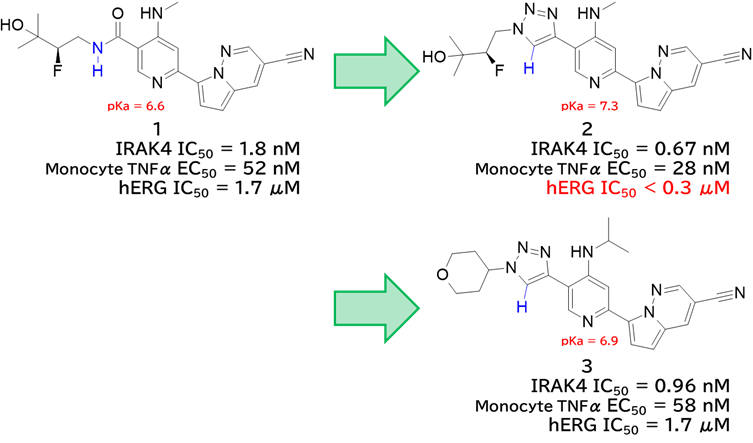
●化合物1はhERG阻害が見られたため、その低減を目指した
・ 先行研究でピリジン部分の塩基性とhERG阻害の相関が示唆されていた
✓ アミド基をヘテロ環に置き換えて塩基性(pKa, 計算値)を下げられないか?
→ 水素結合ドナーNHはヘテロ環の極性化CH-O相互作用で代替
・ トリアゾール2は阻害活性が維持された(むしろ向上)
✓ しかしピリジンの塩基性が強まり、hERG阻害も増強
→ 置換基を変えて塩基性を弱めた化合物3はhERG阻害が減弱
・ 化合物3のトリアゾールをオキサジアゾールに変換した化合物4は更に塩基性が下がりhERG阻害が減弱したが、活性も減弱
✓ チアジアゾールに変換した化合物5は活性が向上
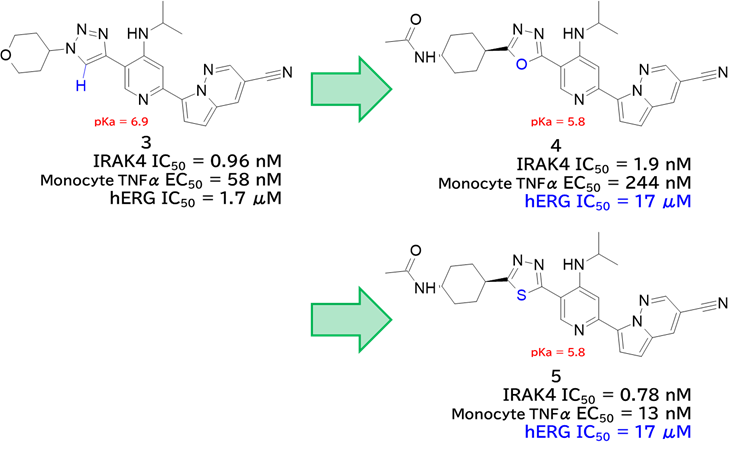
●化合物5-IRAK4複合体のX線結晶構造解析
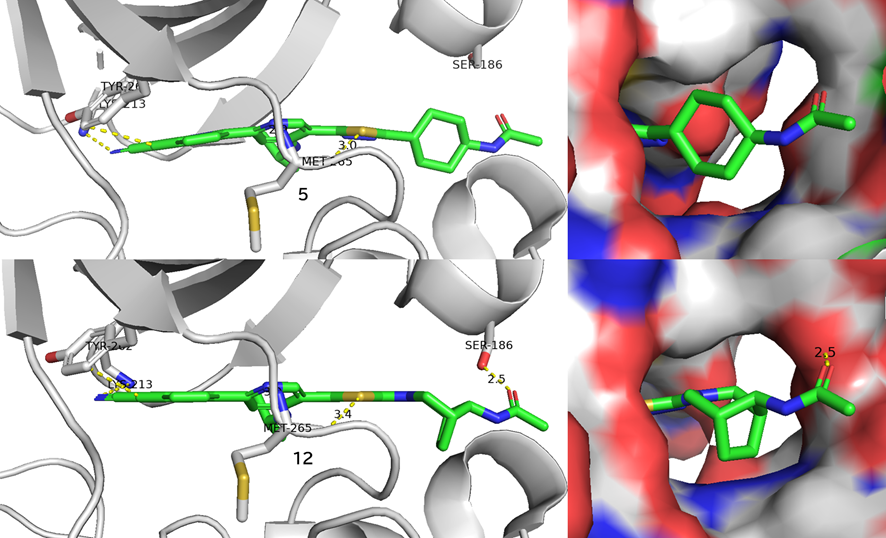
・ 活性向上の一因:チアジアゾールSとMet265主鎖COにS-O相互作用(約3.0 Å)
✓ 硫黄と酸素のファンデルワールス半径の和(3.32 Å)より短い
✓ B3LYP/6-31G**/PBF(water) で DFT 計算を行い、静電ポテンシャルマップと双極子モーメントから、チアジアゾール環の C–S 結合周辺の電子受容性(C–S σ* 方向)を推定
→ S-O相互作用の方向と一致
・ ピロロピリダジン3位シアノ基がLys213残基と水素結合
・ ピロロピリダジンがTyr262残基とT字型(edge‑to‑face)CH-π相互作用
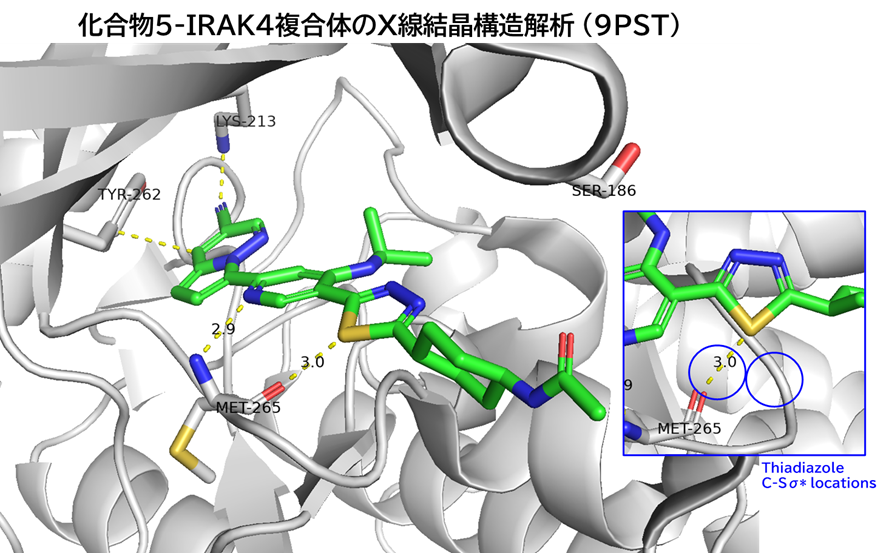
●化合物5をベースに合成展開して化合物22を取得
・ Ligand lipophilicity efficiency (LLE)を指標に合成展開
・ 化合物5のシクロヘキサンを架橋型ピペリジンに変換した化合物12はLogDそのままでEC50が3倍以上改善
→ イソプロアミノ基を2-シアノエチル基に変換した化合物22はLogDを下げつつEC50が6倍以上改善
・ 化合物22はキナーゼ選択性あり(一番狭いIRAK1でも16倍あり)&良好なin vivo PKプロファイル
・ 残念ながらin vivo薬効評価の記載はなし
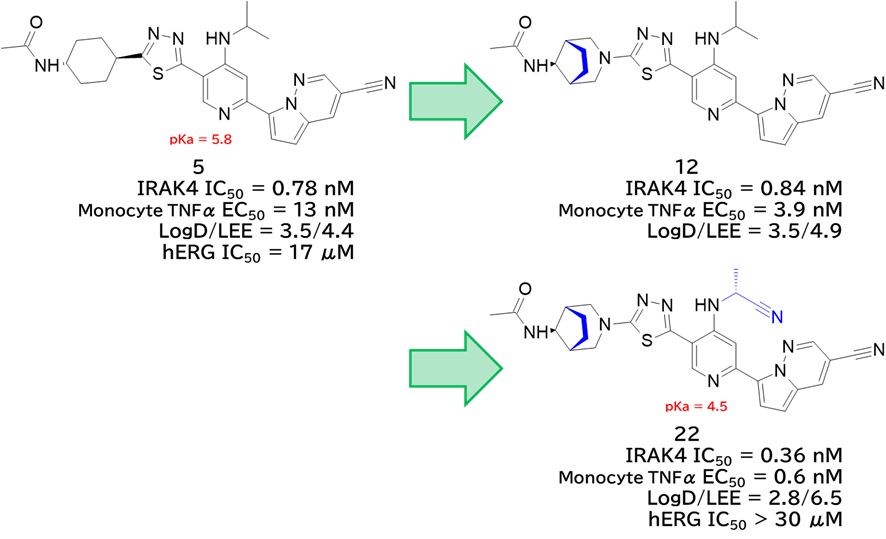
・ 化合物12-IRAK4複合体のX線結晶構造解析から、化合物5と同様の結合様式を確認
・ 新たにアセトアミド基とSer186残基の水素結合あり
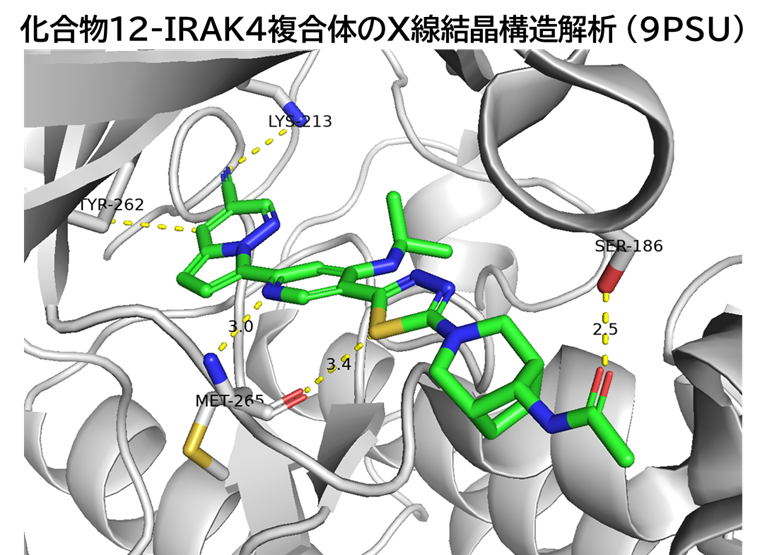
・ 化合物5と12の比較
✓ 化合物5のシクロヘキサンはチアジアゾールと垂直に配置
✓ 化合物12の架橋ピペリジンがチアジアゾールと同平面上に配置
→ 架橋メチレンが疎水性面と接触 (N265、R273、L277 の主鎖が連続した水素結合ネットワークを形成し、その背後に水や極性リガンドがアクセスしにくい疎水性面を作り出している)
✓ アセトアミド基の位置は両者同様なのに、なぜか化合物12はSer186と水素結合を形成
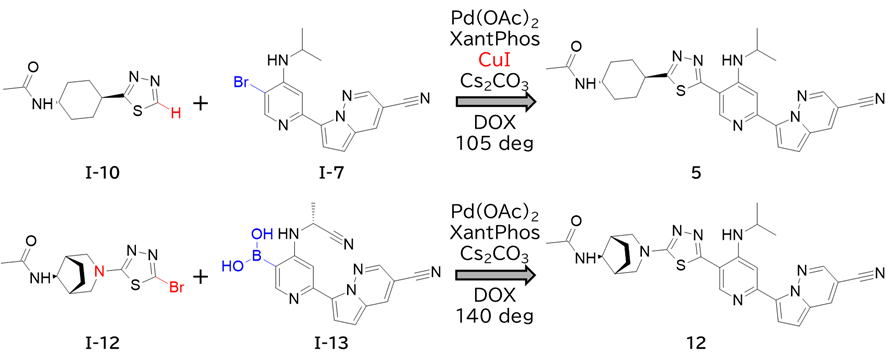
●合成で面白かったポイント
・ チアジアゾールとピリジンのカップリング
✓ シクロヘキサンの場合はC-HとBrでPdとCu触媒のクロスカップリング
✓ ピペリジンの場合はBrとBでPd触媒のクロスカップリング
→ 電子リッチ/プアによって使い分け
【④Accent社のDHX9阻害剤でのS-O相互作用】
Discovery of ATX968: An Orally Available Allosteric Inhibitor of DHX9
J.Med. Chem., 2025, 68, 9537–9554.
https://doi.org/10.1021/acs.jmedchem.5c00252.
●DHX9 は RNA/DNA ヘリカーゼに属し、転写・翻訳の調節や R‑loop の解消、DNA 修復を通じたゲノム安定性の維持に不可欠な多機能酵素である。腫瘍細胞ではしばしば DHX9 の発現亢進や機能異常が報告されており、DHX9 を阻害すると R ループ蓄積、複製ストレス、DNA 損傷応答の破綻が誘導され、腫瘍細胞が選択的に脆弱化することが示されている。こうした知見から、DHX9 は新規な抗腫瘍標的として注目を集めており、特に小細胞肺がんや MSI‑H/dMMR 大腸がんなどで依存性が示唆されている。
そこでAccent社は、腫瘍細胞の脆弱性を突く新たな抗がん剤として DHX9 阻害剤の取得を目指した。
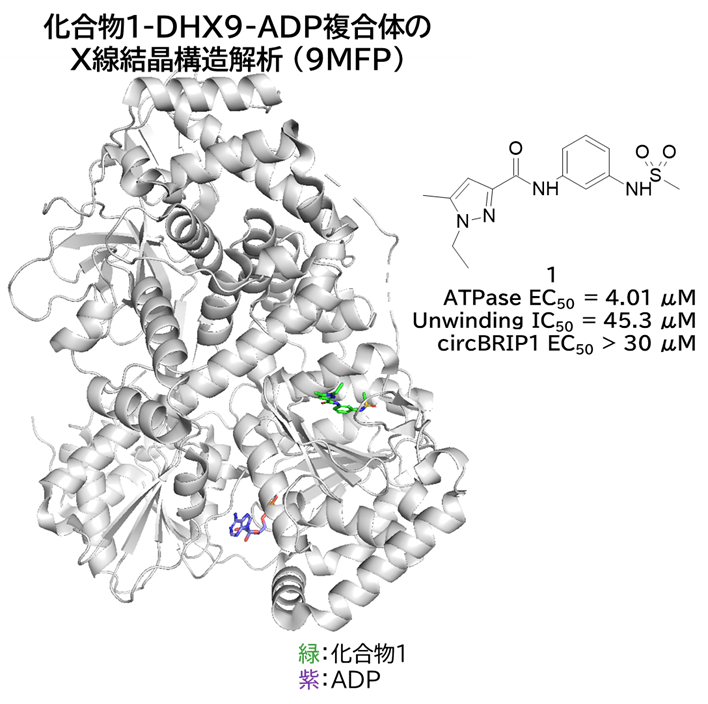
●HTSでヒット化合物1を取得
・ ATPase活性は部分阻害(最大70%)だが、アンワインド活性(ATP を使って二本鎖の核酸を一本鎖にほどく)は完全阻害
・ X線結晶構造解析で、ATP結合部位とは異なるアロステリック部位への結合を確認
✓ 結合部位(アロステリック部位)はヘリカーゼファミリーで保存性が低く、高選択性期待
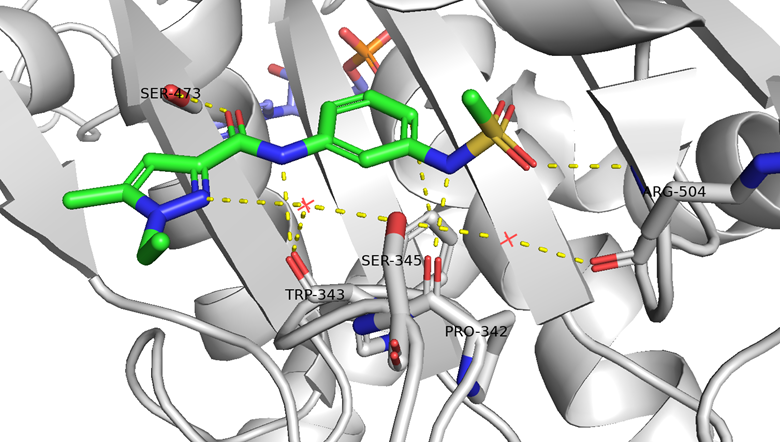
・ ピラゾールNが水を介してSer345残基, Arg504主鎖COと水素結合
・ アミドNHがTrp343主鎖COと水素結合
・ ベンゼンがTrp343残基とT字型(edge‑to‑face)CH-π相互作用を形成
・ スルホンアミドNHがSer345主鎖COと水素結合
・ スルホニルOがArg504主鎖NHと水素結合
●化合物1をベースに合成展開してATX968を取得
・ 上記構造解析より、アミド、ベンゼン、スルホンアミドは活性に必須と示唆(変換が難しそう)
・ ピラゾールをチオフェンに換えて活性が大幅に向上
・ チオフェンのメチル基をピリジンに換えて活性と物性が改善
・ ベンゼンにクロロ基を導入して活性が向上、ATX968を取得
✓ ハロゲン(Cl/Br)の導入はresidence timeを延長させ、細胞活性(circBRIP1 EC50)を向上させた
→ ATPase IC₅₀よりもresidence timeの方が細胞活性と強く相関(R²: 0.55 vs 0.84)
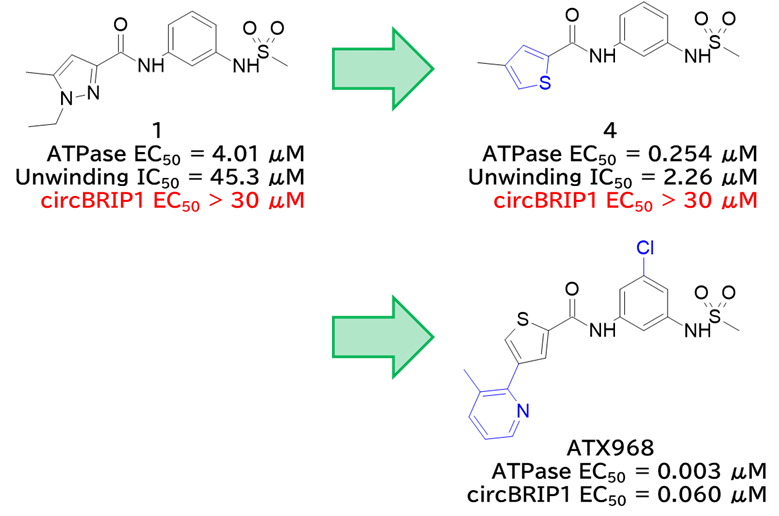
・ ATX968-DHX9-ADP複合体のX線結晶構造解析から、化合物1と同様の結合様式を確認
・ チオフェンはピラゾールのNとは異なる配置
✓ 反転してSer472, 477残基と水素結合
→ それぞれ距離が3.7, 3.4 Åで硫黄と酸素のファンデルワールス半径の和(3.32 Å)より長いのでS-O相互作用では無さそう
→ むしろATX968アミドのカルボニルCOとS-O相互作用して活性配座に固定してそう
・ ベンゼンのクロロ基がCys489と硫黄-ハロゲン相互作用、Val441とv.d.w.相互作用
✓ これが活性向上およびresidence time延長の肝
・ ATX968のプロファイル
✓ 高い選択性(DHX36, SMARCA2, WRN など他ヘリカーゼを阻害しない)
✓ マウスで良好なPK(改善されたクリアランス・AUC)
✓ 300 mg/kg BIDで24時間EC90を維持し、MSI-H CRCゼノグラフトモデルで腫瘍体
今回注目したいのは、2点です。
一つ目は、S-O相互作用の有用性です。
どの事例でも活性向上に寄与しています。水素結合やπ–カチオン相互作用より弱いが、π–πスタッキングや疎水性相互作用より強い、これは利用したいです。そもそもS-O相互作用は、タンパク質内でメチオニン–セリンやメチオニン–スレオニン、ジスルフィド–酸素間を介して安定化に寄与する相互作用のようですが、今回の4例を見た感じでは、硫黄原子はチオエーテルのような電子豊富な状態よりも、チアゾールやチアジアゾールのような電子欠乏した環境の方が相互作用を形成しやすいように思えます。X線結晶構造解析やGiliad社のDFT計算から、硫黄原子の左右斜めの位置(C-S σ*)に3.2 Å以内の距離に酸素原子が配置されるような設計が良さそうです。
また、他の使い方として、例えば環状ペプチドやPROTACのカメレオニシティに利用して物性と細胞膜透過性を両立させるテクに使えないかなとも思いました。
東大 菅先生、京大 後藤先生が環状ペプチド内にチアゾールを入れたりしていた気がしますが、水中だと水と水素結合しつつ、細胞膜内だと分子内S-O相互作用を形成して透過性が上がったりするかも。

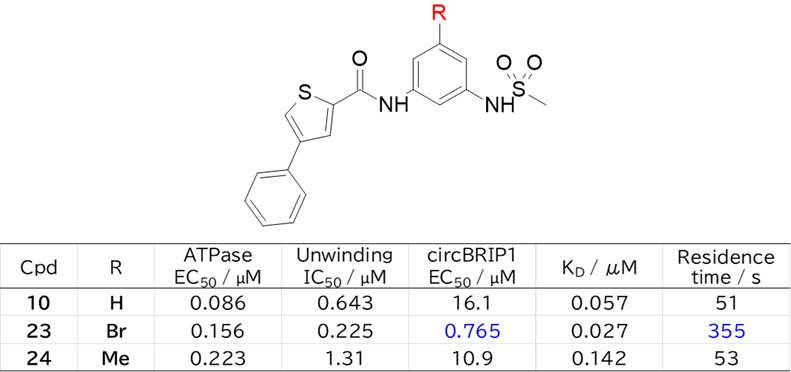
二つ目は、Ascend社の硫黄-ハロゲン相互作用によるresidence time延長です。
もう少し論文を詳しく紹介すると、以下のように無置換10、ブロモ基23、メチル基24でATpase活性に大きな差はありませんが、細胞活性(circBRIP1 EC50)は大きく差が出ています。構造解析から、ハロゲンはCys489と硫黄-ハロゲン相互作用、Val441とv.d.w.相互作用していることが示唆されており、メチルでもv.d.w.相互作用は形成するでしょうから、Cys489との相互作用が肝なのか、硫黄-ハロゲン相互作用という形式が肝なのか・・・。方向性を持った相互作用が重要、ということなのかも・・・?
いやぁ、メドケムって本当にいいものですね。