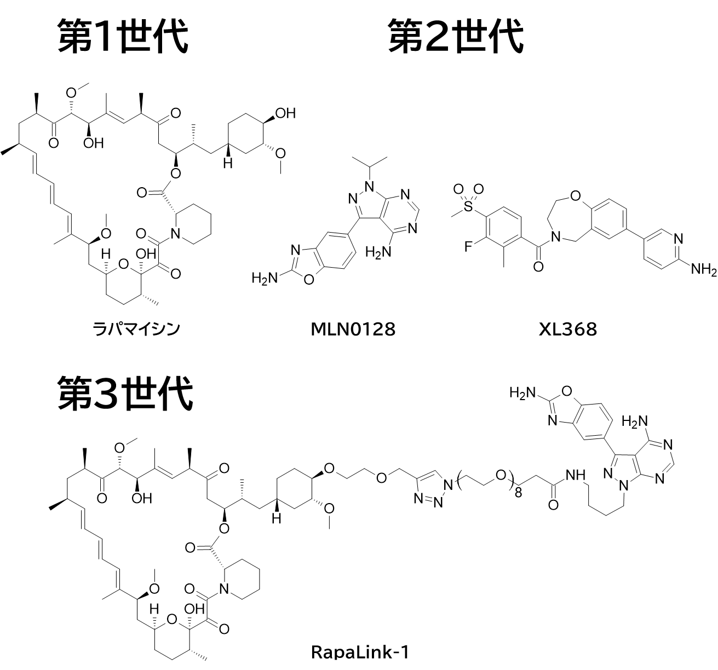
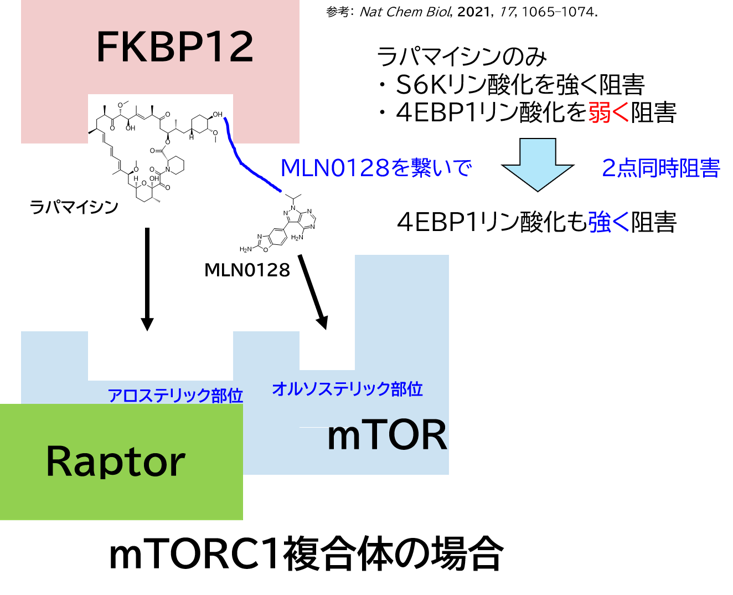
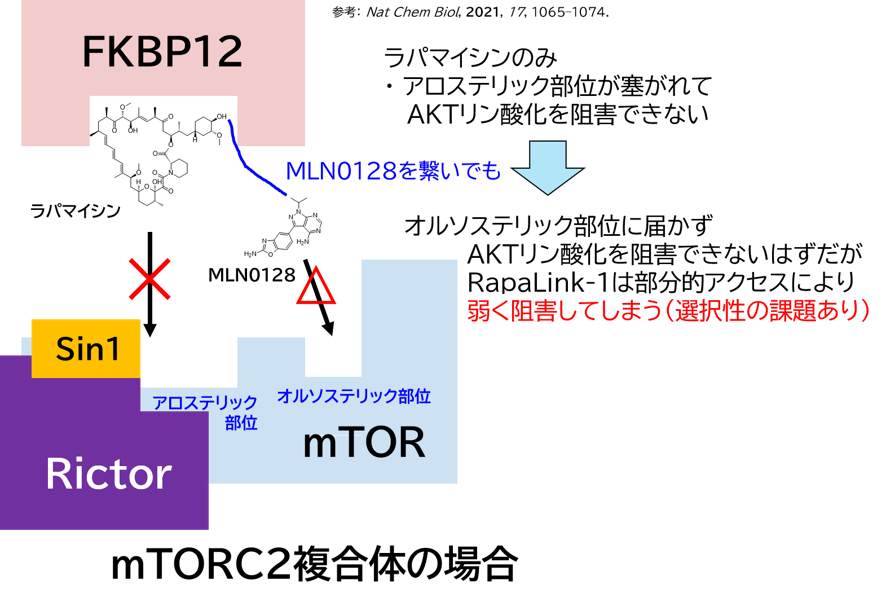
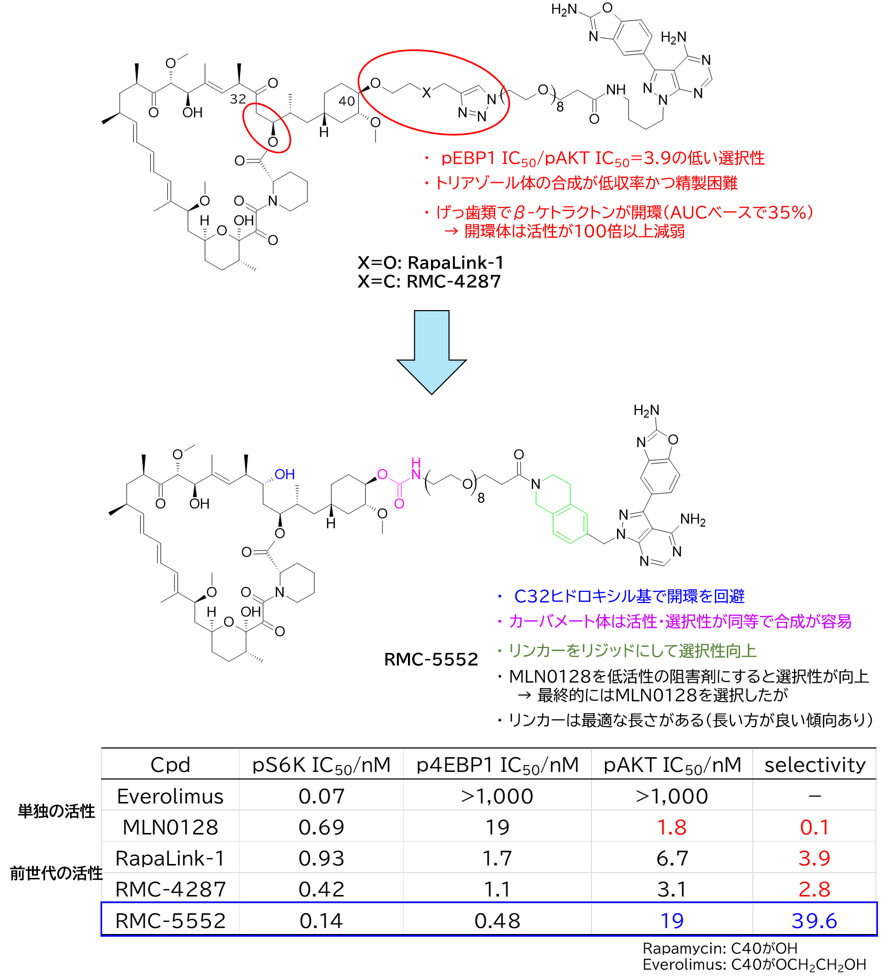
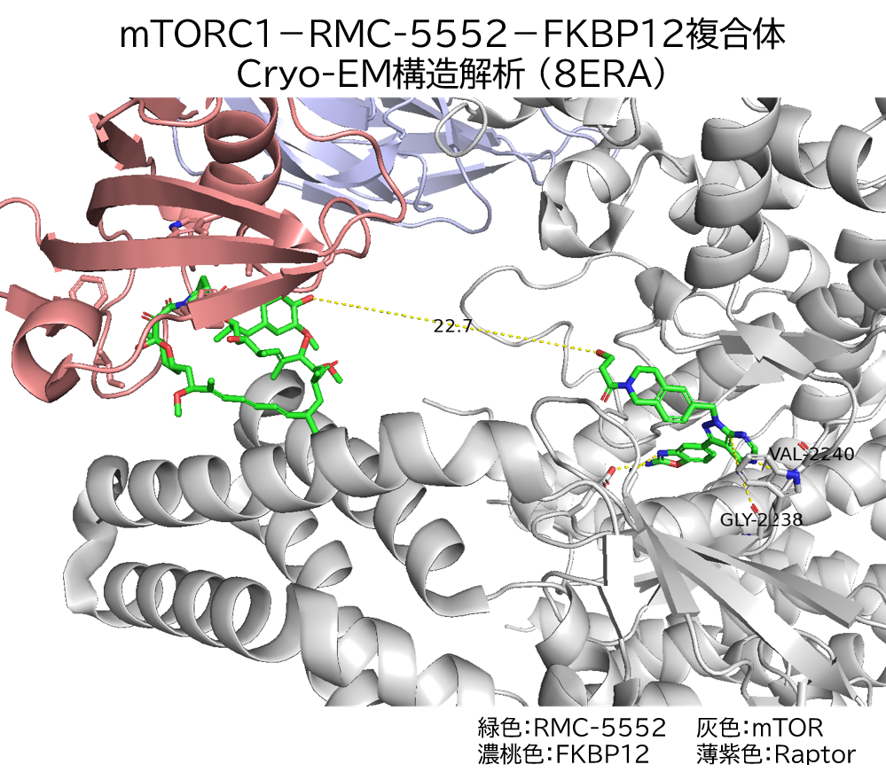
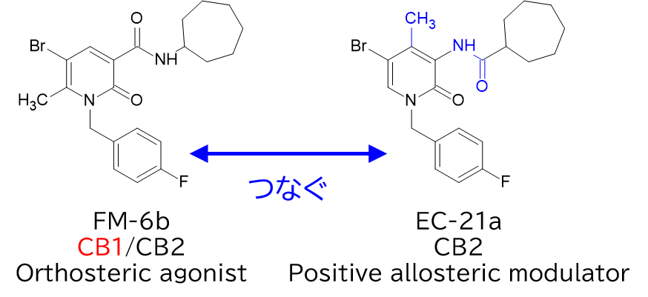
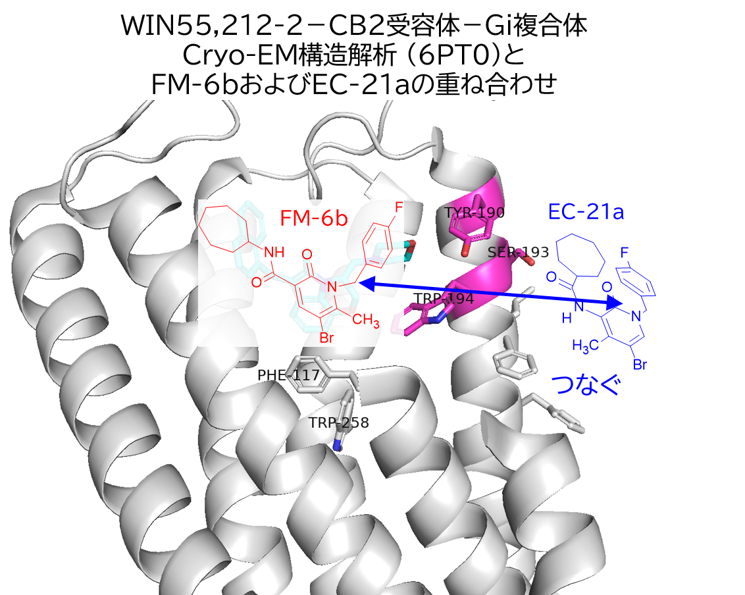
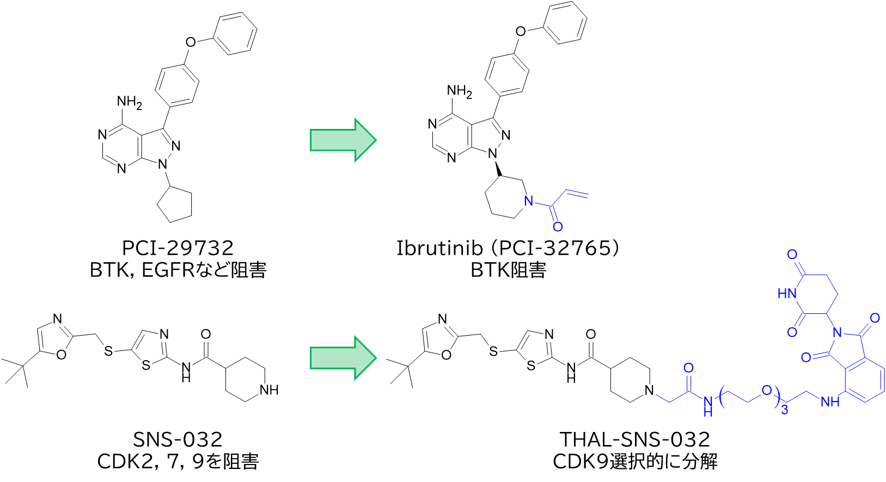
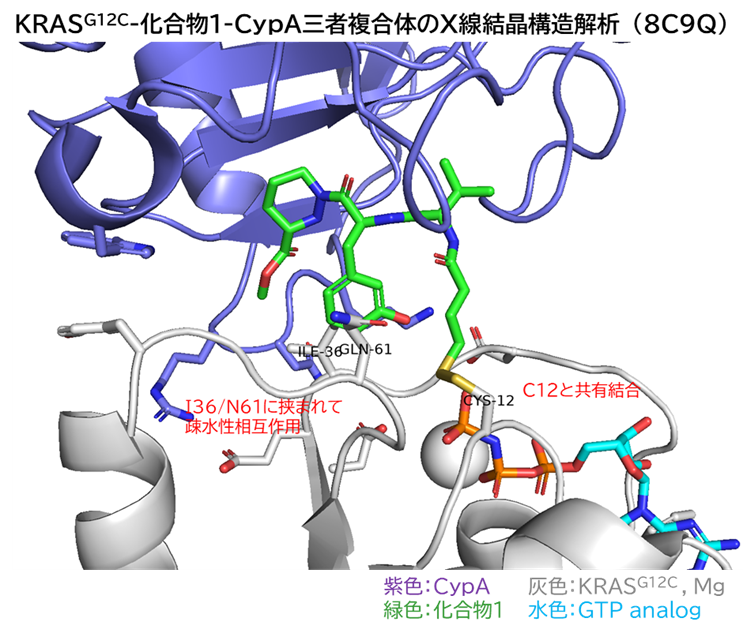
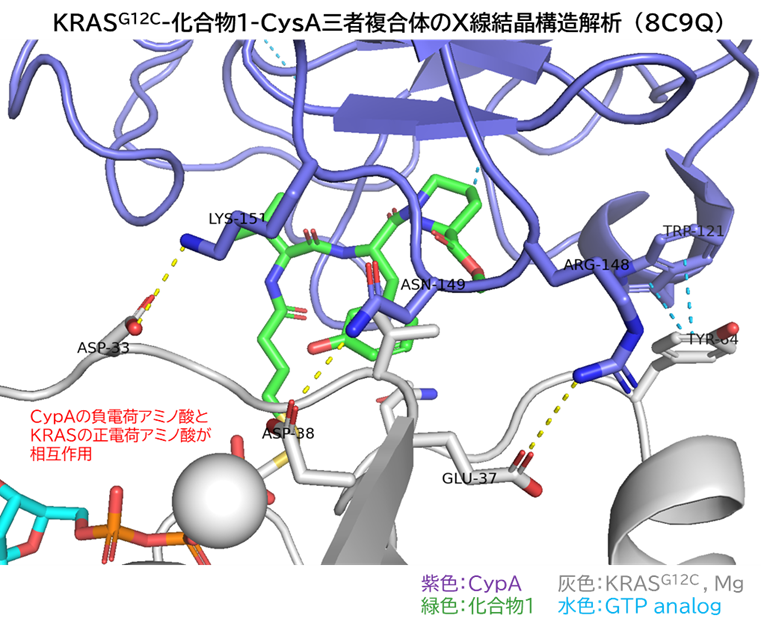
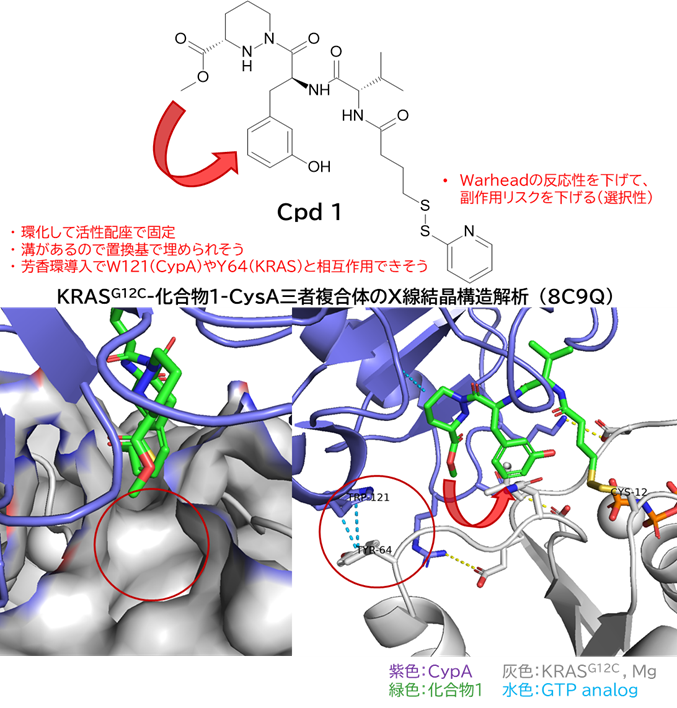
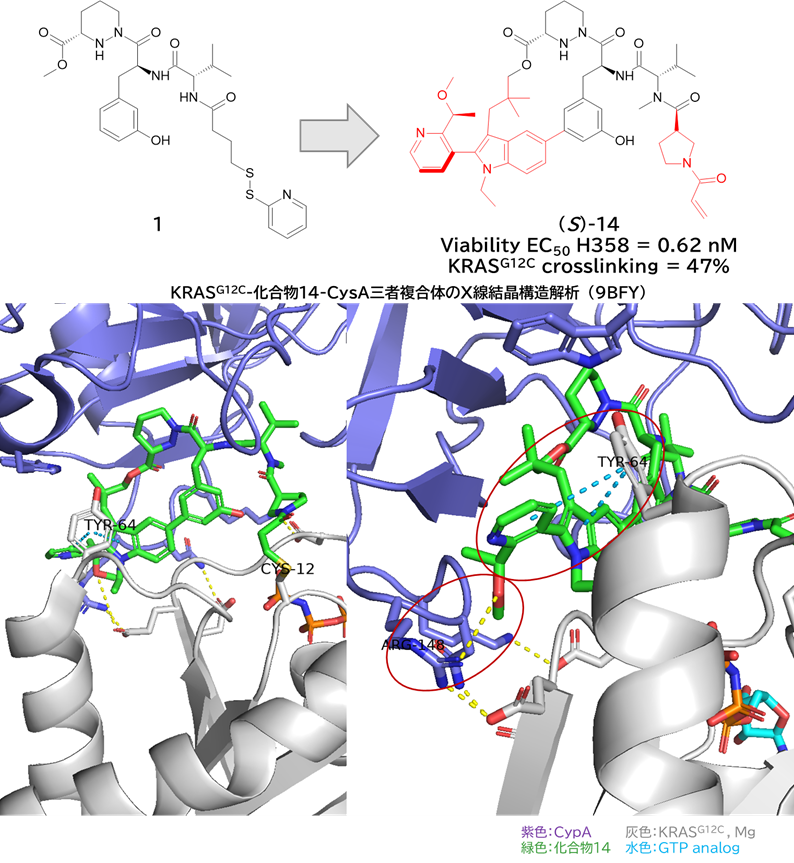
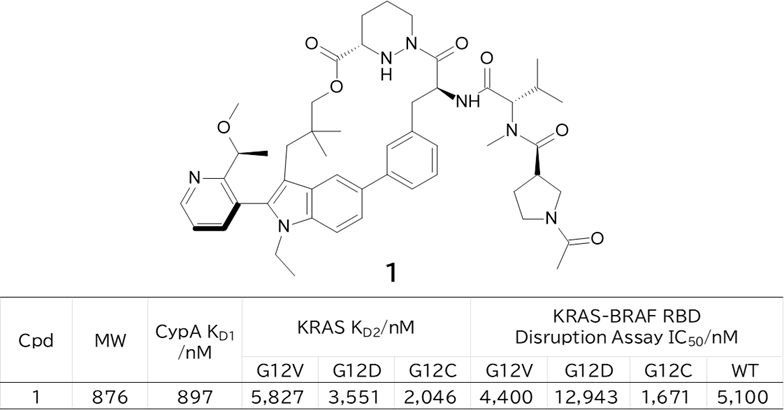
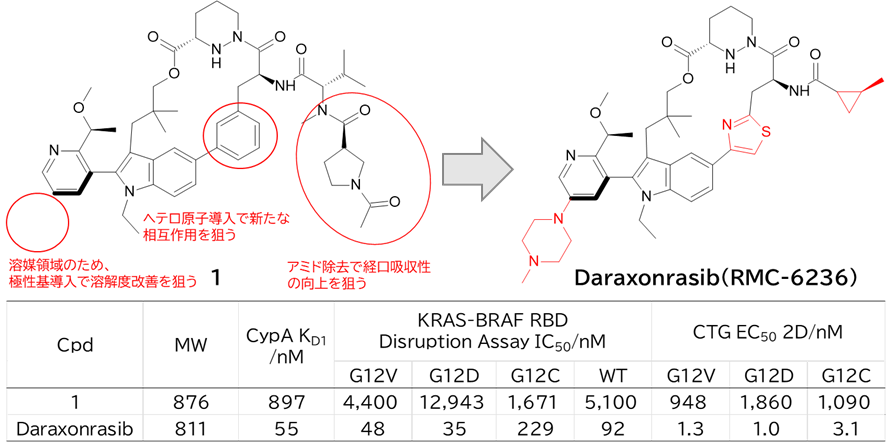
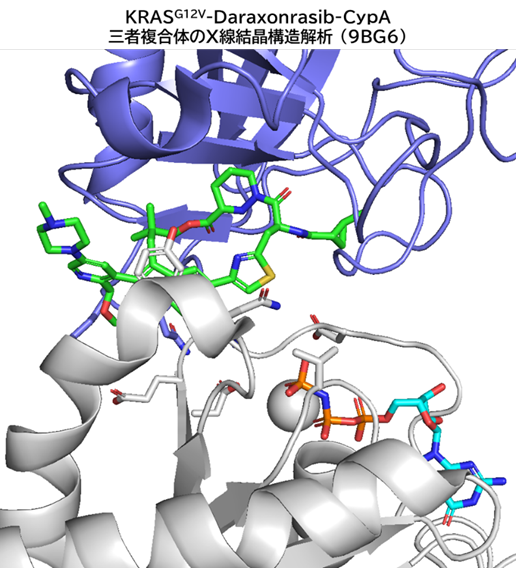
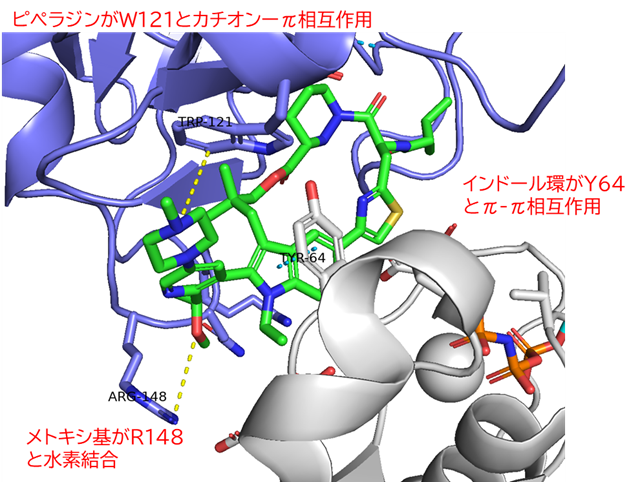
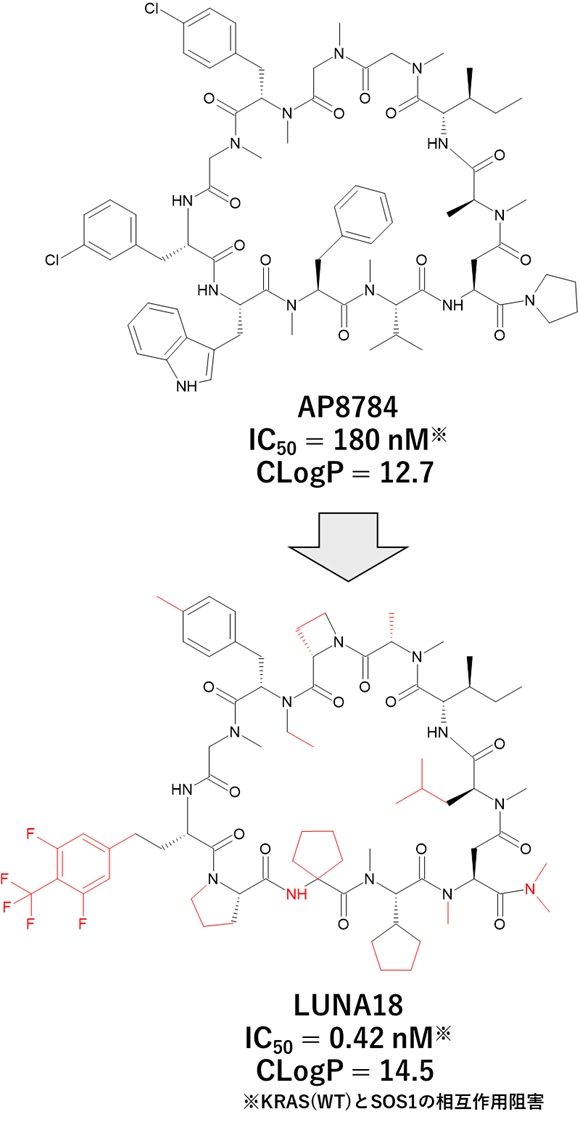
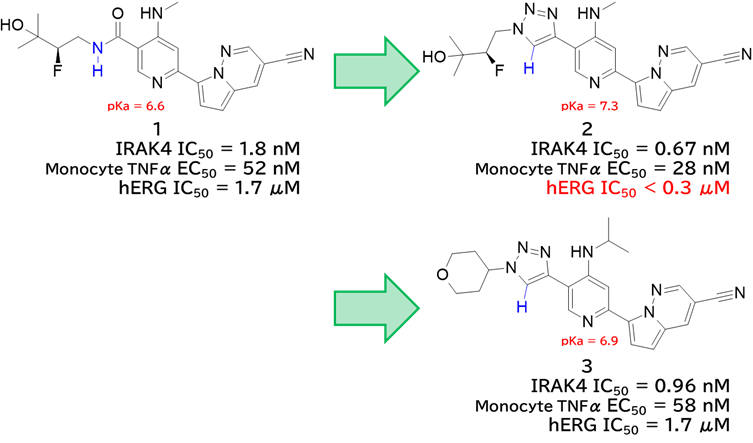
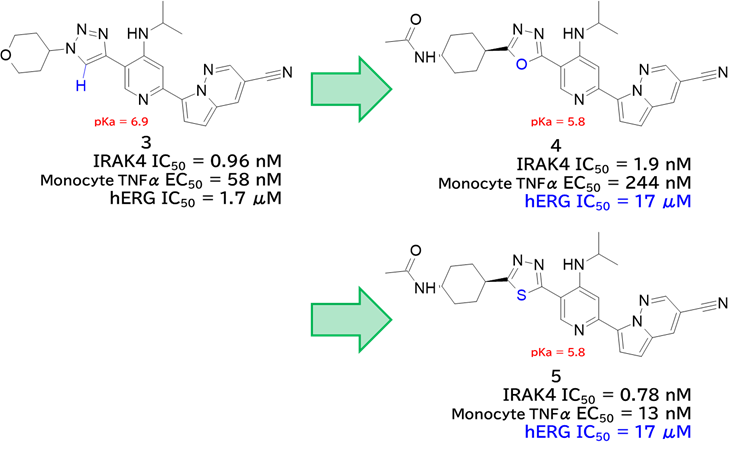
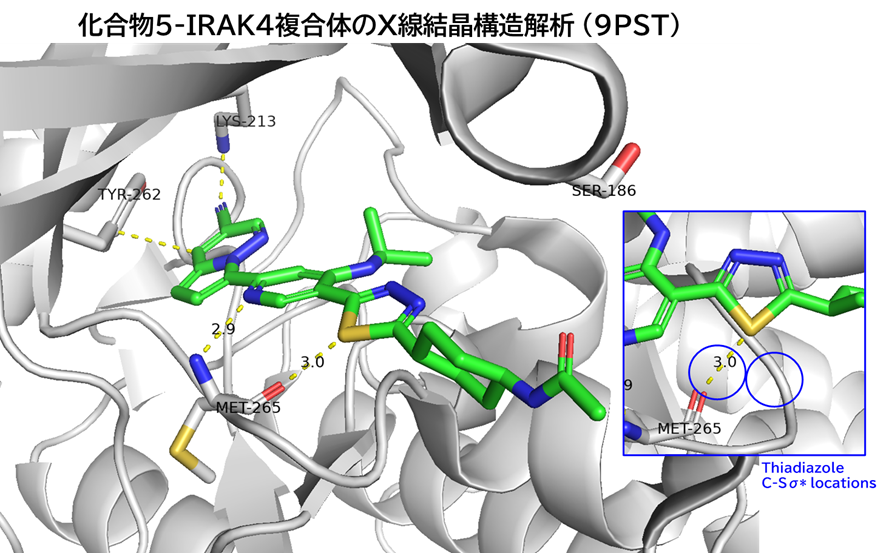
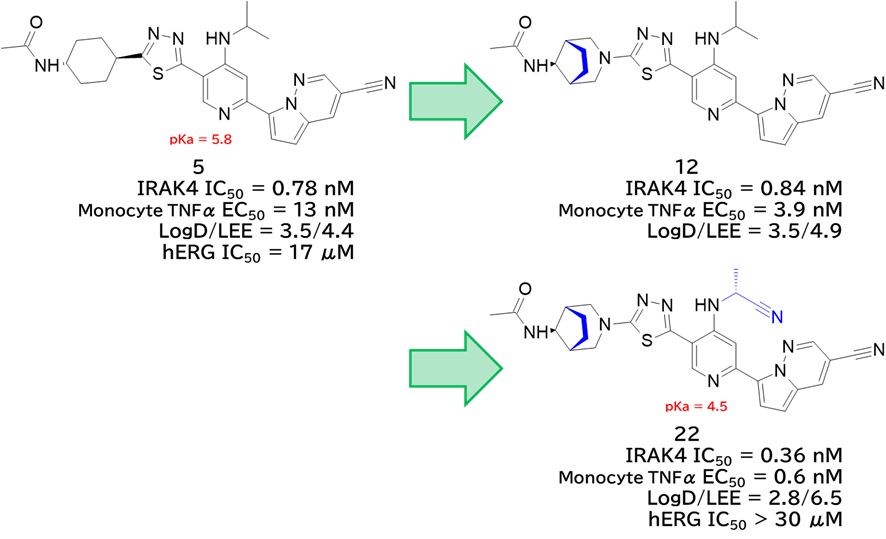
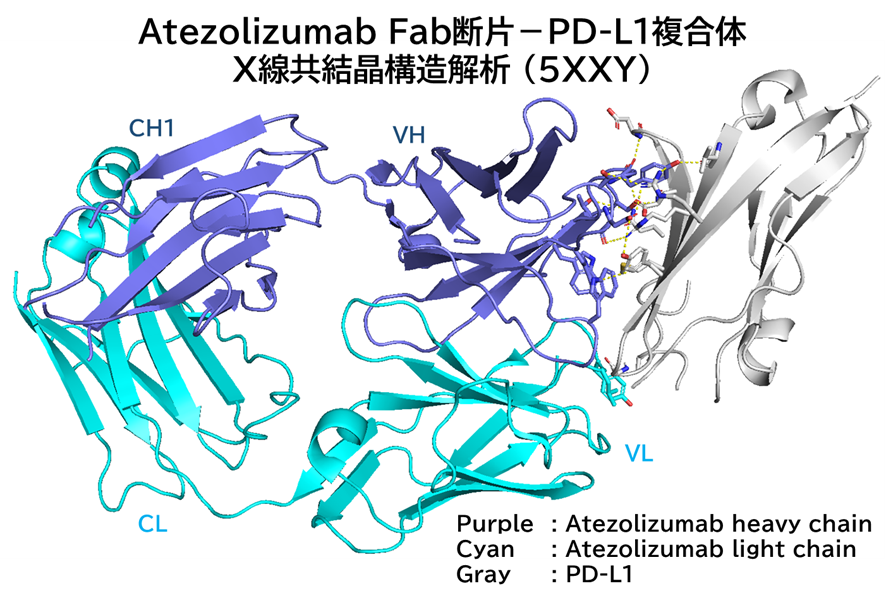
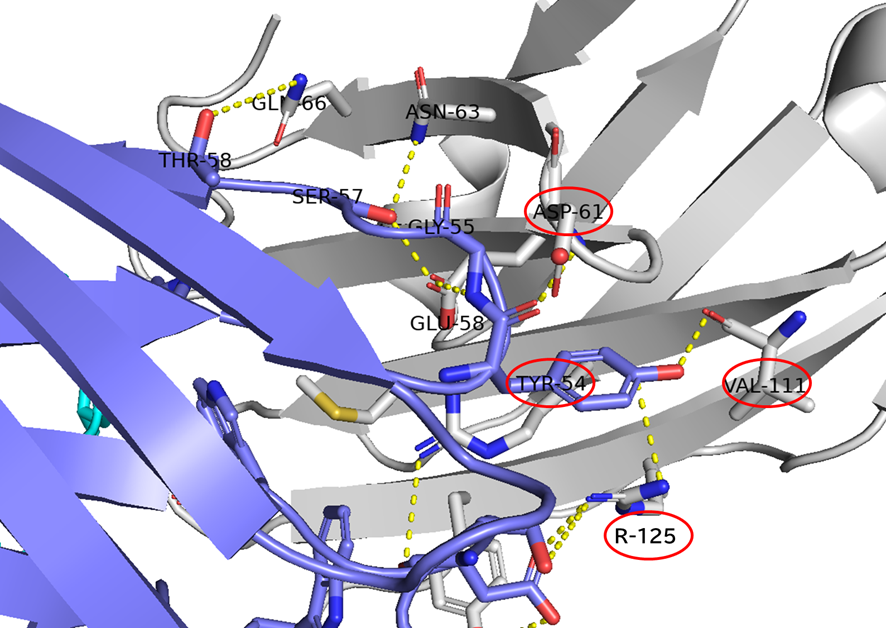
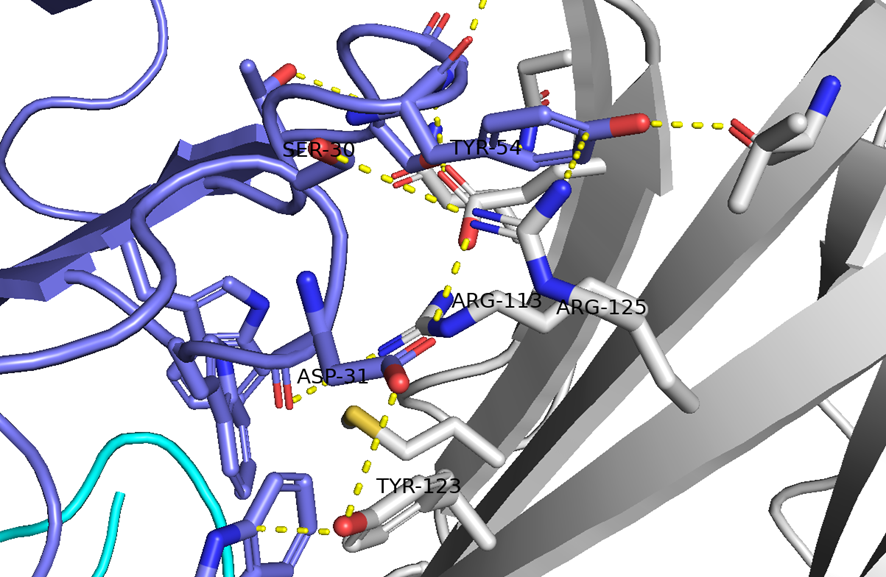
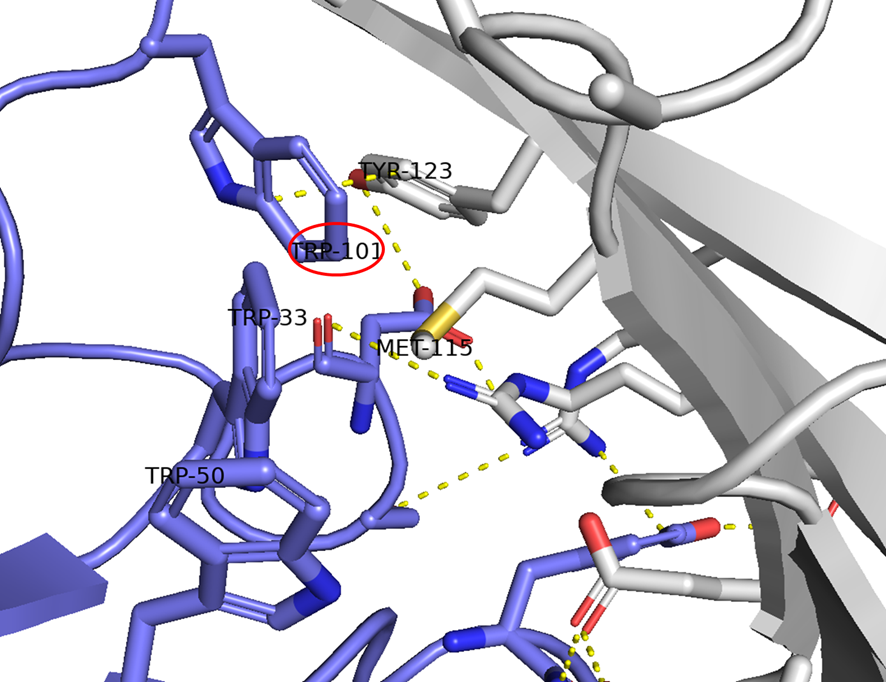
GO CENTRAL(ちょっと長文過ぎた・・・)
【2026/06/26追記】
中分子を脳移行させるには?
PROTACは分子内水素結合でHBDを隠すデザインが鍵。
核酸はDDS必須で、hTfR1ペプチドを使うなら塩基性アミノ酸+トランスフェリンと競合する部位への結合がBBB通過に効く可能性がある。
と書いたけど、そうとも限らないかもしれない。
JCR/Peptidreamの脳移行No.894と、IONIS/Bicycleの筋移行BCY15466は、結合部位・様式が異なり、アミノ酸残基の組合せで作り分けられる可能性がある。
特に脳移行TfRリガンドペプチドは、表面残基を狙えばシミュレーションでde novoデザインできるのでは?
一気に追いついてみせるぞ

【① イントロ: 低分子化合物を中枢移行させる】
4年前の記事『ヘテロ原子が増えて脳内移行性がむしろ改善』から抜粋
https://azarashi-panda.hatenablog.com/entry/2022/05/26/064717
Eli Lily社のZoran Rankovic博士が、市販されているCNS薬とnon-CNS薬の物性の物性を比較している。
CNS Drug Design: Balancing Physicochemical Properties for Optimal Brain Exposure
J. Med. Chem.2015, 58, 2584–2608.
https://doi.org/10.1021/jm501535r
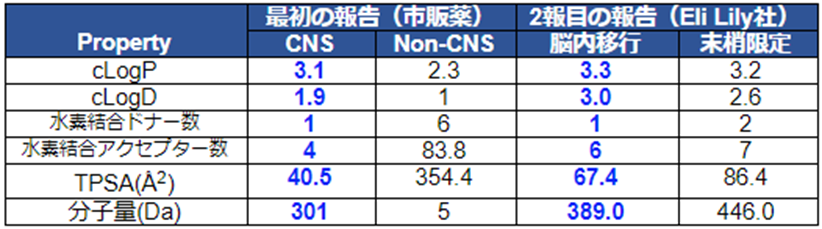
しかし、上記論文において、市販されているCNS薬の89%が伝統的なアミン作動性のGPCRやトランスポーター、イオンチャネルなどを標的とした化合物群で構成され、ケミカルクラスが限定的であった。そこでZoran博士は、Eli Lily社のデータを元にキナーゼやプロテアーゼなどを標的とした脳内移行性化合物(対するは末梢限定化合物)を加えて再解析したところ、CNSを狙う際の化合物の物性(Property Space)は市販のCNS薬よりも広く、重要な因子は分子量と水素結合ドナー数であることが分かった。
CNS Physicochemical Property Space Shaped by a Diverse Set of Molecules with Experimentally Determined Exposure in the Mouse Brain
J. Med. Chem.2017, 60, 5943–5954.
https://doi.org/10.1021/acs.jmedchem.6b01469.
2つの論文の一部を抜粋した表を以下に示す。
また、脳内移行性を妨げる要因の1つである排出トランスポーターP糖タンパク質(P-gp)に関して、Pharma Algorithms社のRemigijus Didziapetrisらは、化合物中に含まれる
・ 窒素原子と酸素原子の数(N + O)≧ 8
・ 分子量 > 400 Da
・ 酸性度pKa > 4
の化合物はP-gp基質になりやすいと述べている。一方で、
・ (N + O)≦ 4
・ 分子量 ≦ 400 Da
・ 塩基性度pKa < 8
の化合物はP-gp基質になりにくい傾向がある。
PROTACやオリゴ核酸のような上記を満たせない化合物はどうだろか?
レビューや論文から探ってみた。
【② PROTACsを中枢移行させる】
『2-1) 最初に、伊トリノ大学Caronらのレビューから現状を把握する。』
BBB-Permeable PROTACs: Where Do We Stand?
ACS Med. Chem. Lett. 2026, XXXX, XXX, XXX-XXX
https://doi.org/10.1021/acsmedchemlett.5c00768.
神経変性疾患との関連が示唆されるタンパク質として、タウやアミロイドβ、α-シヌクレイン、TDP-43らの凝集体、GSK-3βやCDK5、LRRK2のようなキナーゼ、RAGEのような受容体、アポリポプロテインE、プレセニリン1,2 (γ-セクレターゼ複合体の構成成分)などが挙げられ、それらを標的とするには、化合物が脳血液関門 (Brain-Blood Barrier; BBB)を通過して脳組織に到達する必要がある。 しかし、PROTACsのようなルールオブファイブを越えた化合物 (beyond rule of five; bRO5)は、高い分子量、高いTPSA、高いフレキシビリティゆえに、BBB通過性が著しく低い。 そして中枢移行PROTACsのデザイン指標や標準化された評価系は確立途上である。
● BBBの特徴
BBBは脳内皮細胞 (BECs)とアストロサイト、ミクログリア、平滑筋細胞、基底膜などが組み合わさって脳実質細胞を血液から分離して恒常性を確保している。BECにはタイトジャンクションや取込・排出トランスポーターなどが含まれる。
化合物がBBBから取込・排出される経路は主に4種類。
1) 受動拡散
2) キャリア媒介輸送CMT (GLUT1, LAT1など)
3) 受容体介在輸送RMT (TfRなど)
4) 排出トランスポーターEfflux (P-gp, BCRPなど)
加齢や神経変性疾患の影響でBBBが部分的に破綻することがあるが、空間的に不均一であるため期待できない。
● 中枢移行PROTACsを目指すときの課題
一般的に中枢移行を高めるためのメドケム戦略として、①脂溶性 (cLogP/cLogD)を上げる、②水素結合ドナー数 (HBD)を減らす、③極性 (TPSA)を下げる、④分子の剛直性を上げる、⑤イオン化 (pKa)を減らす、の5つが考えられる。
Pfizer社は、それらを総合的に評価する指標CNS MPOを提言している。しかし、CNS MPOはbRo5化合物では予測精度が低下しやすい。分子内水素結合・カメレオニシティのような三次元的・動的な特性の評価が難しいためと考えられる。
中枢移行を予測する評価系は以下の課題がある
【in vitro評価系】
・ BBB評価用にリン脂質を最適化したPAMPA: HTS向きだが、PROTAC吸着、efflux非対応という問題
・ Caco-2 / MDCK-MDR1: 腸管モデルのためBBB予測には不十分
・ iPSC-BEC: ヒトBBBに近く将来有望であり、それを利用したBBB-on-chip (microfluidic)は、生理学的再現性高いが標準化に課題あり
【in vivo評価系】
・ マウスが主流だが排出トランスポーターは種差が大きい。一方、非ヒト霊長類 (NHP)はスケーラビリティや倫理性に課題がある
・ CNS活性を主張するには、unbound Kp,uuの評価が重要だが、多くの研究で未測定
●2017–2025の23報からCNS-targeted PROTAC 30化合物を分析
・ in vitro評価したのは5化合物
✓ 評価系はPAMPAまたは or Caco-2
→ 使われた細胞株の65%が腸管系などnon-CNS由来でBBB透過評価としては限定的
・ in vivo評価したのは 11化合物
✓ LC-MSで脳内濃度が定量されたのは9化合物 (以下に示す)
→ うち6化合物は経口投与
→ うち4化合物は塩基性官能基をもつ
→ うち3化合物は臨床開発中
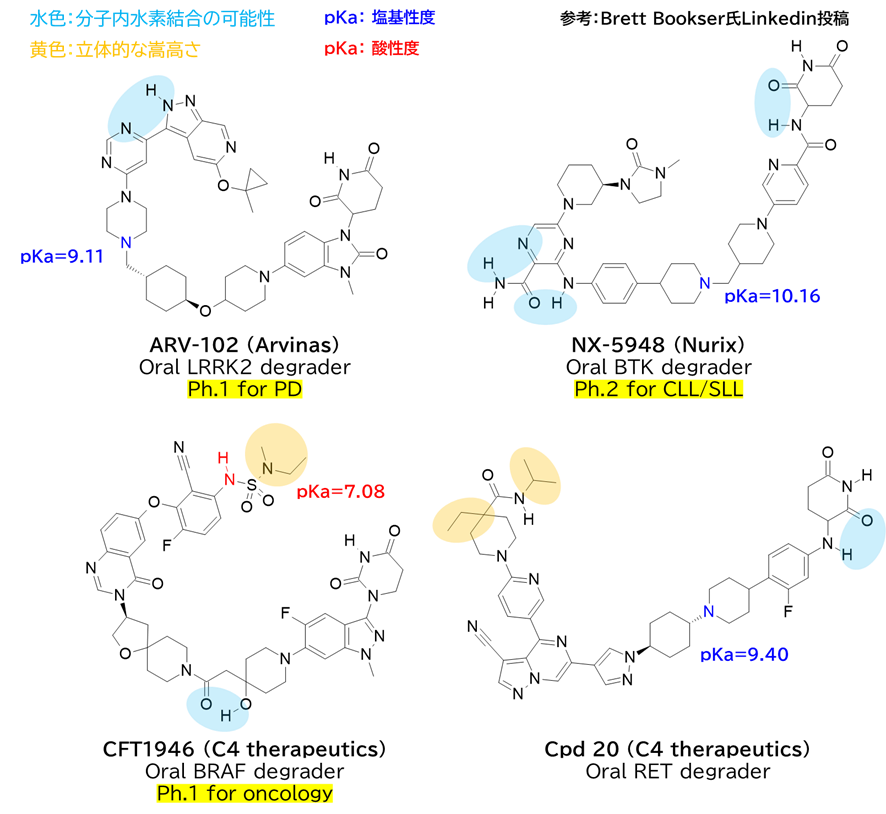
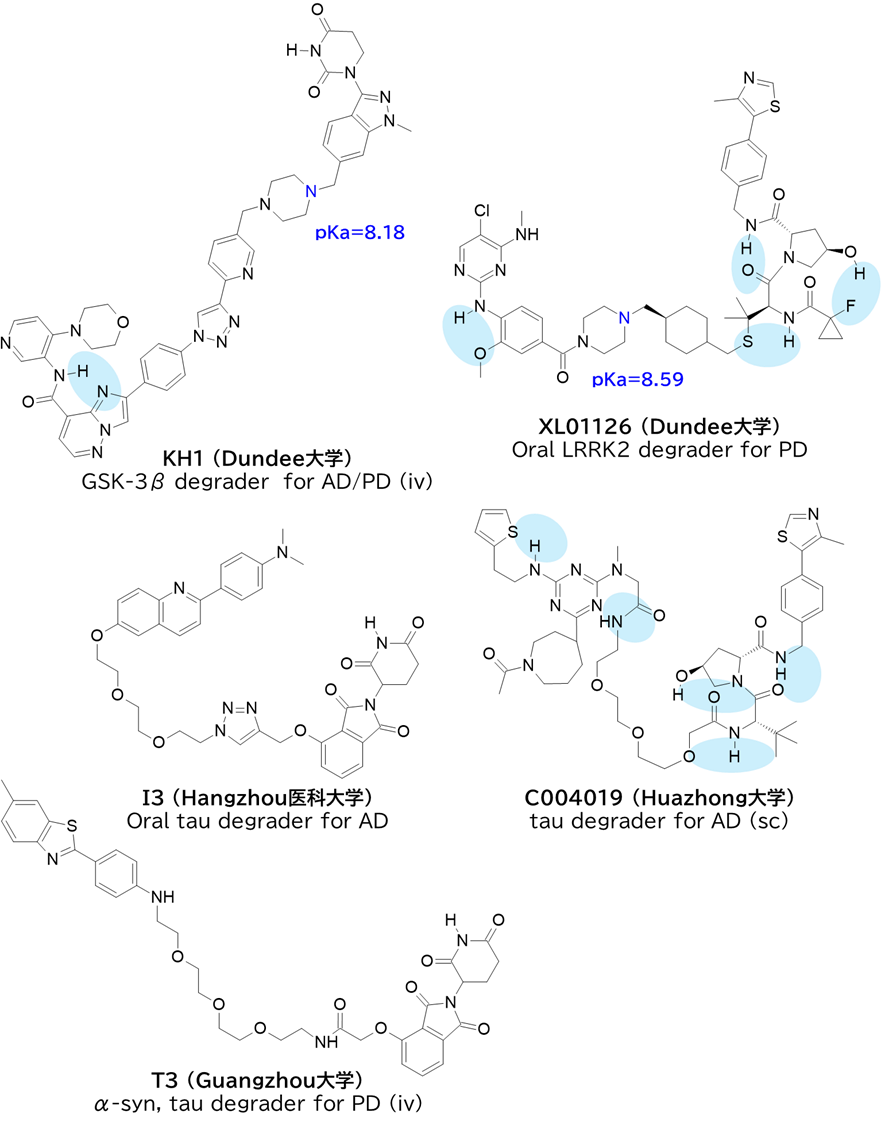
・ 多くが高分子量 (>700), 高TPSA (>160 Ų), 高フレキシビリティ(nRotB >9) に属する
・ E3 ligase: CRBNが多数、VHLは2化合物
・ 水素結合ドナー(HBD)数:2~5個、水素結合アクセプター(HBA)数:14~21個
✓ 分子内水素結合や立体的遮蔽を考慮すると、実効HBDは1程度に低減している可能性
→ CFT1946は酸性官能基(スルホンアミド)を嵩高さで遮蔽している
・ 2D記述子ではCNS-targeted PROTAC 30化合物間の関連性を見出せず
✓ 2D記述子のみではBBB透過性を十分に説明できず、3D構造・動的性質を反映できない
→ 分子内水素結合やカメレオニシティ、RMTなどを考慮した設計が必要
→ AIも必要だが、活用するには高品質で体系化されたBBB透過データが重要だろう
『2-2) 次に、米国ノースイースタン大学のAmijiらのレビューから対応策を考察する。』
CNS delivery of targeted protein degraders
Journal of Controlled Release, 2024, 372, 661–673.
https://doi.org/10.1016/j.jconrel.2024.06.057.
● 分子量の抑制
・ CRBNリガンドは分子量を抑えやすい
✓ CRBNリガンドはVHLリガンドよりも分子量・HBD/HBAが低く、bRo5領域でも物性最適化しやすい
✓ 実際に、CNS-targeted PROTAC(前述)はCRBNが多い
・ リンカーを短くする
・ モレキュラーグルーをデザインする (できれば苦労しない)
● TPSAの削減
・ 分子内水素結合を形成するデザイン
✓ HBDから5,6炭素離れたところに窒素や酸素のHBAを配置
✓ HBDの立体的マスクはP-gp基質認識の回避にも重要
✓ 実際に、CNS-targeted PROTAC(前述)は多く分子内水素結合を形成してそう
・ リンカーのアミド部分をエステルに置換する、または疎水性にする
● フレキシビリティの低減
・ 柔軟性が高いとP-gp基質になりやすいため、マクロサイクル化や剛直化が有効
✓ カメレオニシティはマクロサイクルに有効 (環状ペプチドのシクロスポリンのような)
● pKaの調整
・ 弱塩基性(pKa 6–8)が受動拡散によるBBB通過に有利
✓ Targeted Protein Degraders (TPD) は中性〜弱酸性のものが多く、受動拡散による膜透過性が低くなりがち
→ リンカーの微調整で改善可能
✓ 実際に、CNS-targeted PROTAC(前述)はリンカーに塩基性を持つ化合物が多い
→ そもそもCNS-targetedに限らずピペリジン/ピペラジンとか多いから、中枢移行以前に膜透過性/経口吸収に重要と考えられる
● E3 リガーゼの脳内発現には偏りがある
・ CRBN: 複数の脳領域で安定して発現し、CNS TPDの主要な実績を持つ
✓ 臨床入りしている CNS TPD の多くが CRBN を採用
・ VHL: 脳内発現データはデータベース間で不一致だが、実際には脳内分解の成功例が複数存在
✓ Ubihub (The Ubiquitin Proteasome System Knowledge Base)では主要脳領域でのVHL発現が低い一方、HPA (Human Protein Atlas)では一定の発現が報告されている
✓ tauやLRRK2などでVHL系TPDが脳内で作用した報告がある
・ MDM2, cIAP1: 脳での発現が限定的で、CNS用途では優先度が低い
CNS疾患では標的タンパク質 (POI)の局在・細胞種特異性も複雑であるため、『E3 ligase発現 × POI局在 × 標的脳領域』の三者適合がCNS TPDの鍵
● 投与経路/製剤/DDSの適用
・ Intrathecal(髄腔内)やICV(脳室内)、Intranasal(経鼻)の投与経路は、直接脳へ到達させられるが、侵襲性が高く、均一分布が難しく、長期投与には不向きとされる
・ 受動拡散を高める製剤として、固体分散体やシクロデキストリン包接、脂質製剤(LNP、リポソーム)、ポリマーNP(PLGA)が用いられている
・ 受容体介在輸送RMT
✓ 受容体としてTfR, InsR, LRP1などを利用
✓ 受容体に結合する抗体(IgGやscfv, VHH)やペプチドを付加 (BBB shuttle)
個人的には、TPDは投与経路や製剤、DDSに頼らずに、メドケム的分子デザインによって単剤で経口投与、中枢移行させたい気持ちではある。
とは言え、例えばPPI阻害バインダー+E3 ligaseリガンドのような高分子量が予想されるPROTACにはBBB shuttleが必要かもしれない。ましてAntisense Oligonucleotide (ASO)やsiRNAのようなオリゴ核酸には必須であろう。
それでは、BBB shuttleはどのようなものがあるだろうか?
【③ Brain Shuttle Peptide (脳移行ペプチド)による中枢移行】
『3-1) 最初に、スペインのラモン・リュイ大学Salviaらのレビューから全体を把握する。』
Brain Shuttle Peptideの2015-2025年トレンド
New Trends in Brain Shuttle Peptides
Mol. Pharmaceutics, 2025, 22, 1100–1109.
https://pubs.acs.org/doi/10.1021/acs.molpharmaceut.4c01327
● Blood-Brain Barrier (BBB)が中枢疾患を狙う際のボトルネック
・ BBBは多層的バリア機能をもつ
✓ 物理的バリア: タイトジャンクションによる細胞間隙の制御
✓ 代謝バリア: CYPなど代謝酵素による分解
✓ 輸送バリア: P-gpなどトランスポーターによる排出
・ 一方、BBBはGLUT1など栄養素輸送のためのトランスポーターや受容体も備えている
● BBB Shuttle Peptideによる2種類の脳移行アプローチ
・ 吸着性トランスサイトーシス (Adsorptive-Mediated Transcytosis, AMT)
✓ 塩基性アミノ酸を多く含み細胞膜の硫酸塩などと相互作用する細胞透過ペプチド (Cell-Penetrating Peptide, CPP)であるHIV-TATやR8などを利用した輸送機構
→ 非特異的かつ細胞毒性が懸念
・ 受容体介在性トランスサイトーシス (Receptor-Mediated Transcytosis, RMT)
✓ 脳血管内皮細胞に発現する受容体に結合するリガンドペプチドを利用した輸送機構
1) Angiopep-2: 標的は低密度リポタンパク質受容体関連タンパク質1 (LRP1)
2) ApoE(133-150): 標的は低比重リポタンパク質受容体 (LDLR)
3) T7, T12, THR, B6: 標的はトランスフェリン受容体1 (TfR1)
4) GSH: 標的はグルタチオントランスポーター
5) RVG29: 標的はニコチン性アセチルコリン受容体 (nAchR)
6) RGD: 標的はインテグリン受容体
✓ 脳ペプチド以外のモダリティで報告例のあるRTM
7) インスリン受容体 (INSR)を標的とした抗体
8) グルコーストランスポーター (GLUT1)を標的とした低分子
● BBB Shuttle Peptideの取得・最適化アプローチ
・ 取得法: 天然タンパク質由来の配列を短縮化やファージディスプレイにより取得
・ 最適化法
1) 親和性に重要な部分配列を抽出して短縮化
2) D-アミノ酸化や環状化、ペプチド結合の向きや残基の不斉を反転して代謝安定化
3) 複数のペプチドを付与 (多価化)してエンドサイトーシス促進
4) AMTとRMTを組み合わせて輸送効率を上げる
5) 二次ターゲティング: 脳内の特定部位に特異的に移行するDDSペプチド (ニューロン特異的/アストロサイト特異的/ミクログリア特異的など)や、特定部位に特異的に発現する代謝酵素 (MMP, Cathepsin, Caspaseなど)に切断されるリンカーを追加することで選択性を上げる
● BBB Shuttle Peptideの適応
・ 適応疾患: 膠芽腫やアルツハイマー病、パーキンソン病、筋萎縮症、脳炎など
・ 適応方法: ナノ粒子に多価付与が最も多いが、低分子やオリゴ核酸に直接付与もある
・ ペプチドの抗体と比べて優位性:
✓ 製造容易性や低コスト
✓ 化学修飾の自由度
✓ AMT利用
✓ 組織拡散性や投与経路
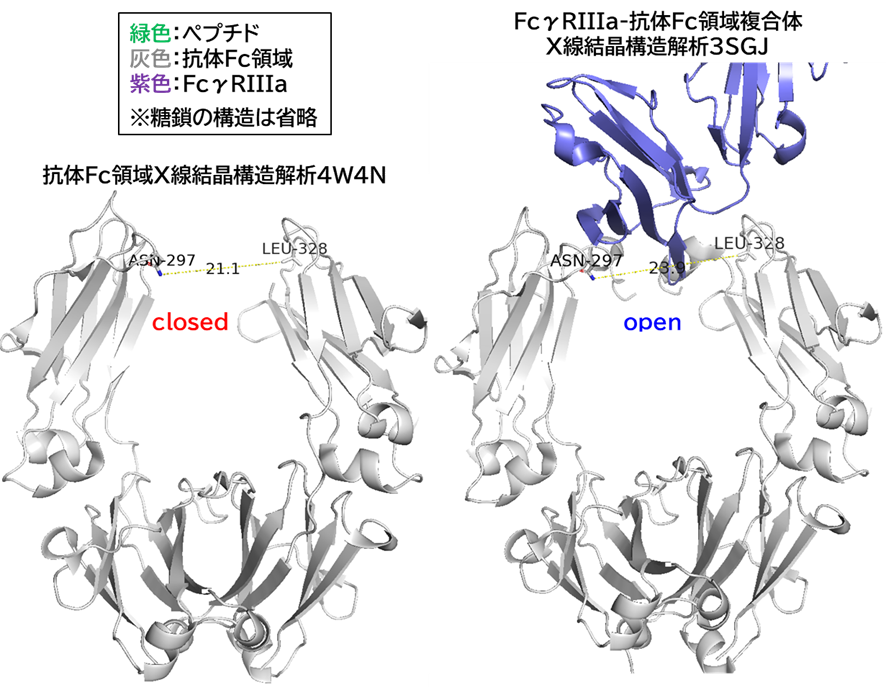
✓多価化: 抗体も多価化はできるが、過剰な親和性はトランスサイトーシス後の脳実質細胞への放出に不利であり、例えば二価 (IgG)だと内皮細胞に強く保持されてトランスサイトーシスしにくく、一価(Fab, scFv)だと BBB を越えやすいらしいので、実際はキビシイだろう
・ 逆に抗体の優位性: 高い結合親和性や血中滞留性 (安定性)、定量・イメージング
これまで同定されているRMTで利用する標的は、BBBだけでなく他組織にも発現しているため、今後はBBB特異的な標的の探索・適用が期待される。またCaronらのレビューでも述べられていたが、BBBに適応した評価モデルの構築が重要と考えられる。
『3-2) 次に、BBB shuttleの事例から特徴を学ぶ』
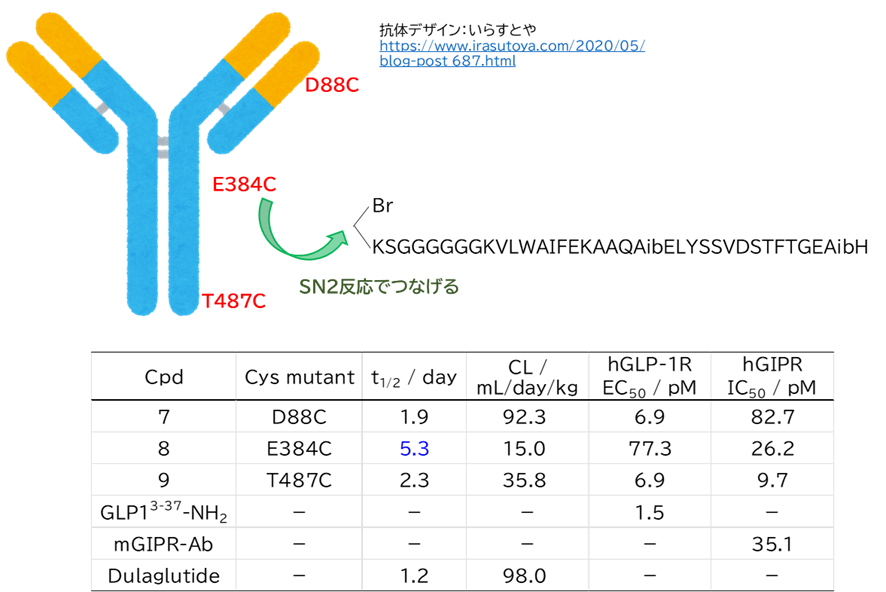
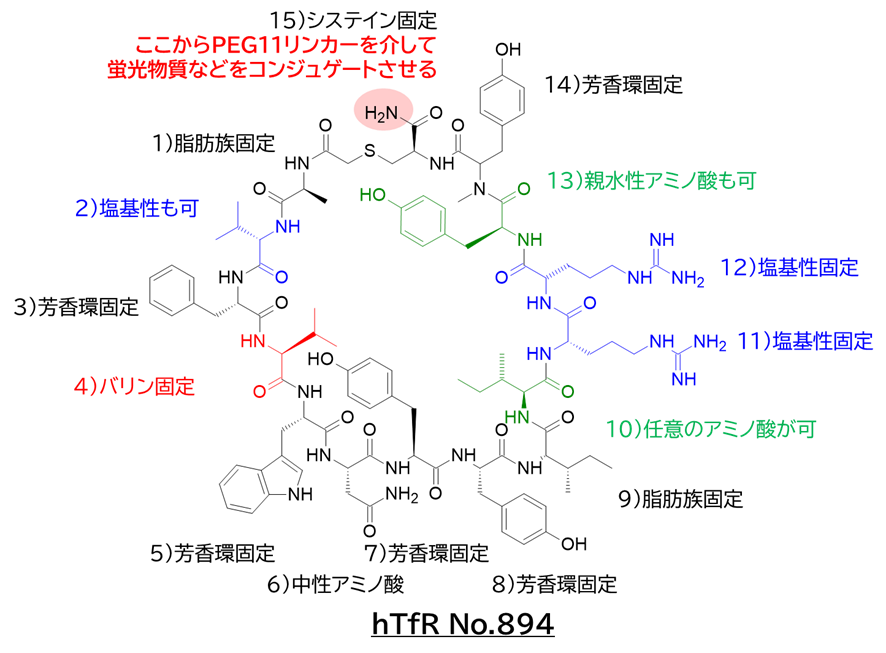
● WO2021167107:ヒトトランスフェリンレセプター結合ペプチド
出願人は、JCRファーマおよびペプチドリーム
ヒトトランスフェリンレセプター(hTfR)に結合することで、血液脳関門(BBB)を通過することができるペプチド,筋組織に指向性を有し、筋組織に効率的に移行することが出来るペプチド及び細胞浸透性を有するペプチド
● マウス脳移行性・脳内局在確認試験
・ hTfR結合環状ペプチドNo.894と蛍光物質のコンジュゲート体
✓ hTfR (SPR) = 0.28~1.09 nM (蛍光物質: FITC, vivotag740)
・ TfR-KIマウスにiv投与
✓ ネガコン投与群と比べて胸椎、心臓、大腿骨、大腿四頭筋および脳により強い蛍光を観測
✓ コンジュゲート体のBBB通過、小脳内部への導入を確認
→ 複数回投与によってプルキンエ細胞のようなニューロンにまで到達
・ BBBを通過し脳内へ移行すること、および筋組織を中心とした各組織へ移行することが確認された。
・ 別で、ヒト乳がん細胞BT-549での試験で細胞内への移行性を確認
・ 以下、No.894の構造および請求項を参考にした必須アミノ酸
● BeBetter社がSOD1*を標的としたsiRNA (siSOD1)とhTfR No.894のコンジュゲート体 (POC2)を評価
* 神経変性疾患(特に ALS)と病態関連性が高い遺伝子で、既報のsiSOD1配列があって比較研究に適しており、siRNAによるKD効果がin vitro / in vivoで評価しやすい
TfR1-Binding Peptide Conjugation Facilitates Robust and Specific siRNA Delivery to the Central Nervous System
Bioconjugate Chem. 2025, 36, 1377–1383.
https://www.sciencedirect.com/org/science/article/pii/S1043180225001223.
・ 大槽内投与 (ICM)および髄腔内投与 (IT)を実施。脂質C16のコンジュゲート体 (C16-siSOD1)とKD部位および作用を比較評価
✓ 両投与経路において、POC2およびC16-SOD1は共にCNS全域でSOD1 mRNAを強いKD作用を示した
✓ POC2は末梢組織でほぼ作用せず、脳内濃度が肝臓の3倍で、28日後までKDが持続
✓ 一方、C16-siRNAは脳内だけでなく抹消組織も強くKD作用を示した
→ 脳内と肝臓で同等の濃度 (ICM/IT投与したsiRNAが抹消に移行するのか!?)
→ C16の高脂溶性に依ると考えられる
・ POC2リンカーの検討
✓ 長さの調整:[2-(2-aminoethoxy)ethoxy]acetic acid/AEEAをスペーサーとして入れたり入れなかったり
✓ 電荷を変える検討: 中性のSar×10、両性のAsp-Lys-Asp、塩基性のArg×3
→ 長さや電荷はKD活性に影響なし(以前にkiwiさんが毒性に影響するのでは?とコメントしていたが、毒性やiv投与でのKDへの影響も気になる)
hTfR No.894は、特許でBBB透過性が示されていたが、それだけでなく論文により、CNS選択性・持続性・低末梢暴露を兼ね備えていることが示された。また、C16もCNSへの作用として強力で有用だが、ICM/IT投与であっても抹消に作用するリスクが考えられた。
ところで、hTfRリガンドは、脳内移行だけでなく、筋肉組織への移行で使用されることもある。以前にバナナさんから親和性の違いでコントロールできると教えていただいたが、特許でのNo.894と蛍光物質のコンジュゲート体のhTfR KD値は、0.28~1.09 nMと非常に高く感じられた。他に何か特徴的な違いはないだろうか?
『3-3) 最後に、他組織を標的としたhTfRリガンドと比較する』
● Ionis社とBicycle社のTfR1結合ペプチドによるオリゴ核酸の筋肉・心臓に選択的な移行
Conjugation to a transferrin receptor 1-binding Bicycle peptide enhances ASO and siRNA potency in skeletal and cardiac muscles
Nucleic Acids Res., 2025, 53, gkaf270.
https://doi.org/10.1093/nar/gkaf270
・ TfR1結合二環性ペプチドBCY17901を同定し、ASO/siRNAにコンジュゲートさせることで筋肉・心臓へのデリバリー効率を大幅に向上させた
✓ BCY17901は、ASO/siRNAの筋組織でのED50を5〜9倍改善し、心臓でも同等の改善を示した
✓ サルおよびhTfR1 KIマウスに対して、骨格筋・心筋における強力なKD作用を示した
・ BCY17901–ASOを全身投与しても、脳皮質・脊髄でKDが見られず、BBBを通過しないことが示された
✓ BBBでのTfR1介在性トランスサイトーシスには、より低親和性のTfR1リガンドが必要
✓ hTfR KD (SPR) = 7.8 nM (BCY17901単独), 5.6 nM (Dmpk ASO conjugate), 22.7 nM (Malat1 ASO conjugate), 42.9 nM (Hprt siRNA conjugated)
→ 出元が異なるので比較できないが、No.894の方が数字上は高親和性・・・
● BCY17901取得の経緯
・ ファージディスプレイでhTfR1に対する二環性ペプチドスクリーニングを実施
・ トランスフェリンと競合しないヒットペプチドBCY15466, 15468を取得
hTfR1発現細胞に対して蛍光ラベル化ペプチドで内在化・局在化を観察
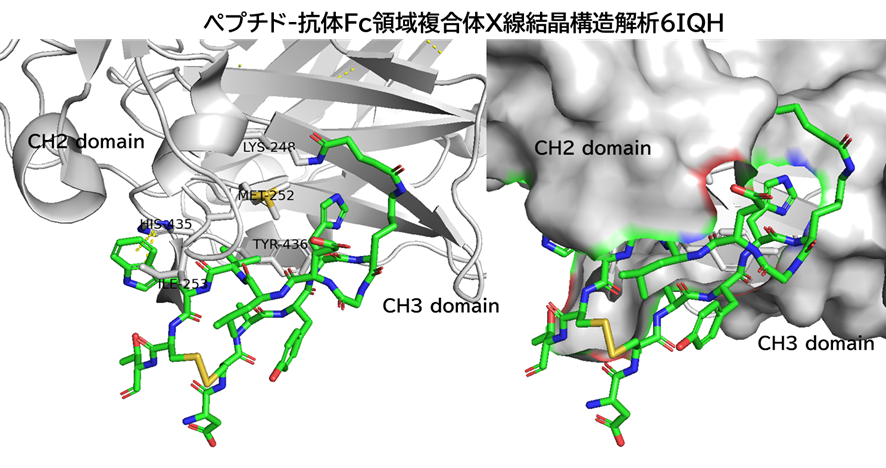
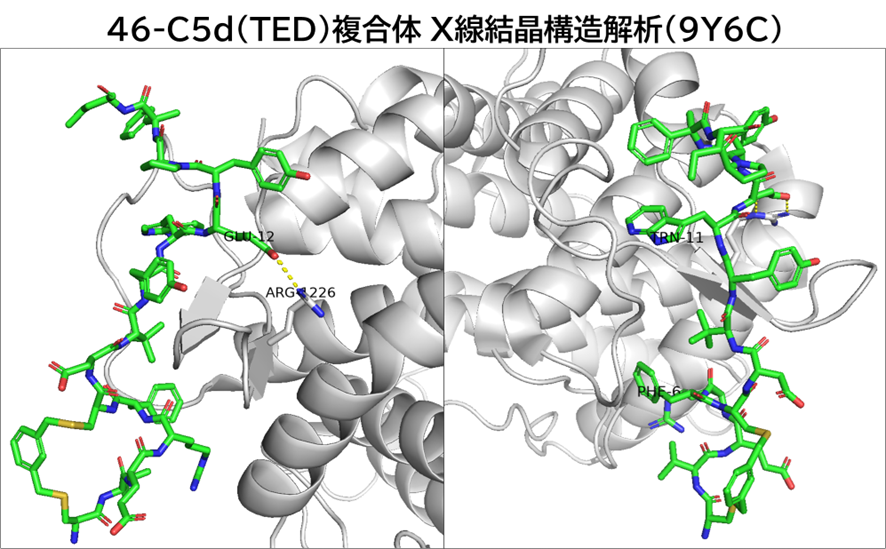
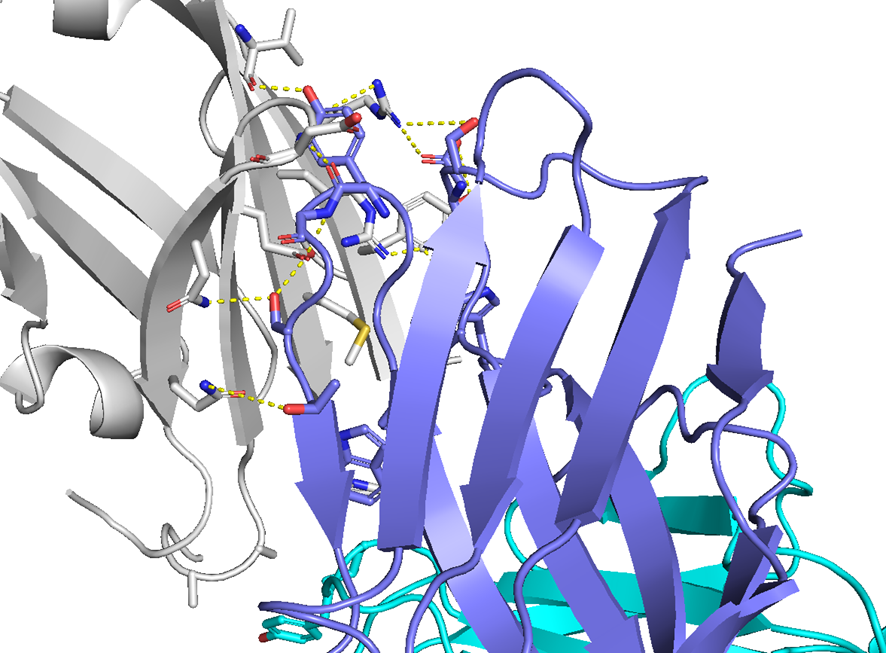
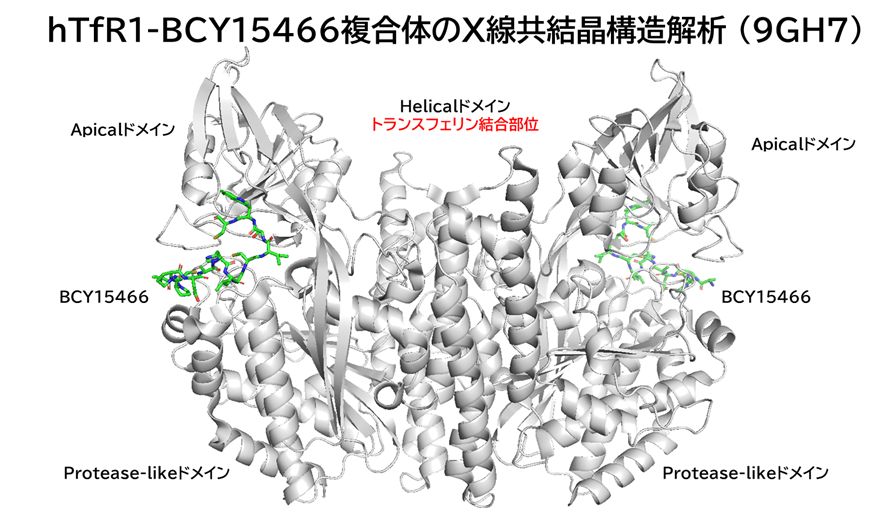
・ BCY15466とhTfR1の共結晶を取得、X線構造解析を実施
✓ BCY15466はトランスフェリンとは異なる部位(Apicalドメイン)に結合

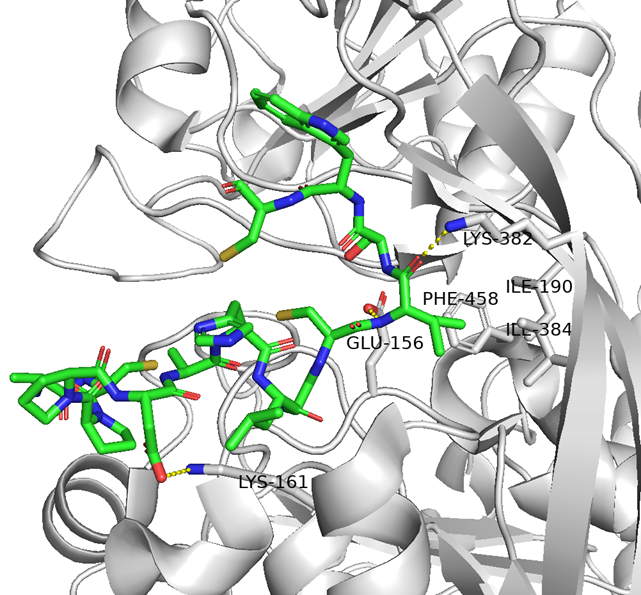
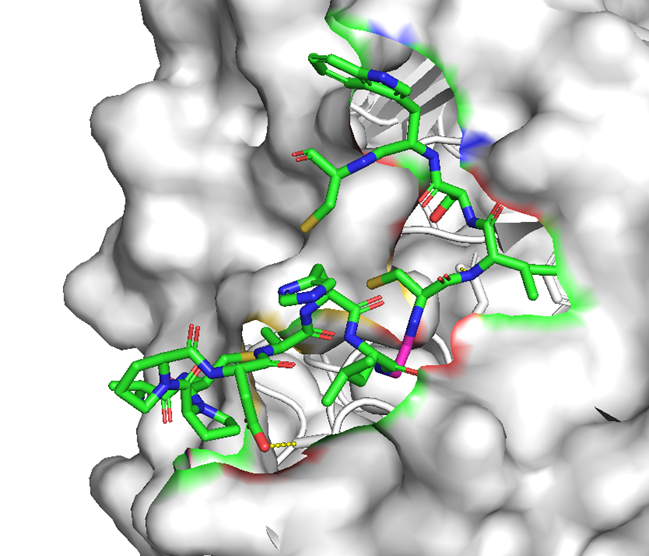
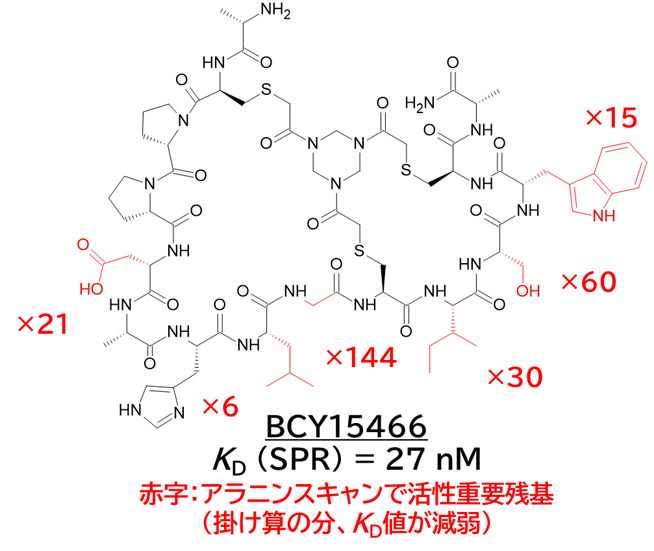
・ BCY15466のアラニンスキャンで結合親和性に重要な残基を把握
✓ 前述のX線構造解析と同様の結果
✓ 合わせてメドケム展開を行い、BCY17901を取得
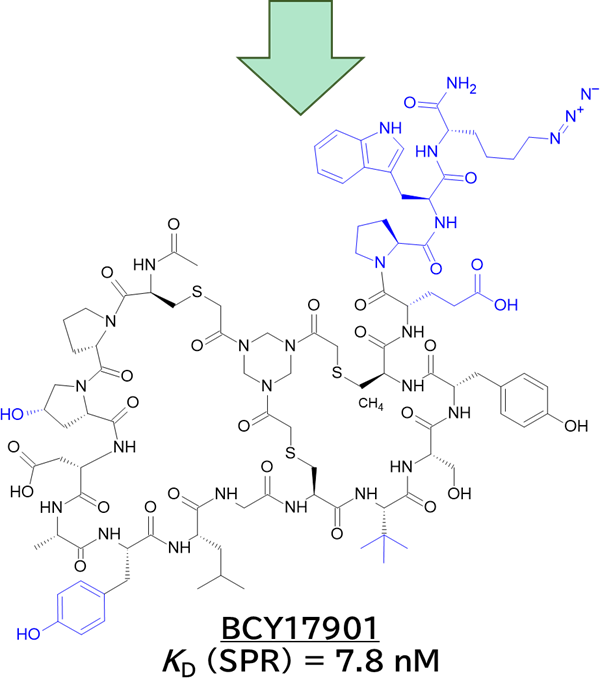
ここで注目したいのは、No.894とBCY17901の比較です。
前者はTfR1介在性トランスサイトーシスでBBBを越える一方、後者はエンドソーム内に取り込まれるものの、結合が維持されたままリサイクリング経路に乗って細胞外へ戻されると考えられます。ここで「BBB通過には、より低親和性のTfR1リガンドが必要」と言われていますが、特許や論文のSPRのKD値を見る限り、数字上はNo.894の方が高親和性に見えます。この矛盾は、KDの絶対値以外の要素が影響していると考えられます。
一つの違いとして、No.894には塩基性アミノ酸(Arg)が含まれています。これはCPP(TAT, R8など)でも知られるように、エンドソーム膜との相互作用や膜不安定化に寄与し得るため、エンドサイトーシス後に部分的なエンドソーム脱出を促し、結果として BBB 通過に寄与した可能性が考えられます(と言ってもNo.894にArgは2つしか入っていないけど)。つまり、TfR1リガンドに限らず、BBB shuttleペプチド全般において塩基性アミノ酸が重要になるかもしれません。
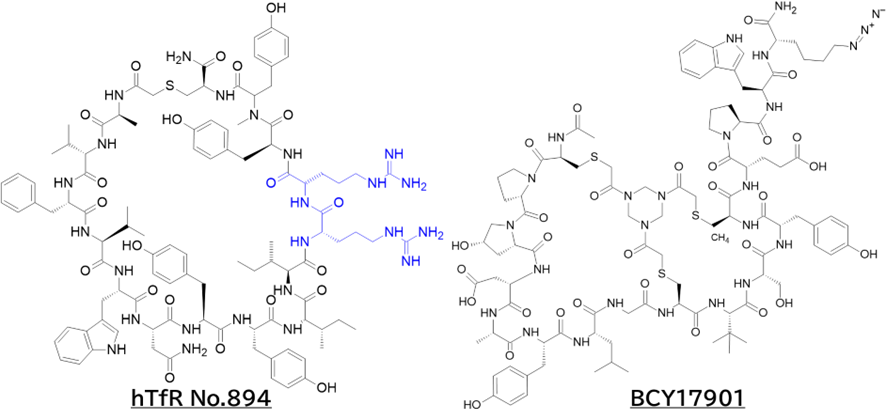
さらに妄想レベルではありますが、BCY17901は疎水性相互作用や水素結合を中心としたinduced fitにより結合が安定し、解離が遅いためリサイクリング経路に留まりやすい一方、No.894はイオン性相互作用が多くinduced fitが起こりにくい可能性が考えられます。また、BCY17901がApicalドメインに結合することが分かっているのに対し、もしNo.894が(構造情報はないが)Helicalドメインやトランスフェリン結合部位近傍に結合しているとすれば、トランスフェリンの結合・解離サイクルに巻き込まれてトランスサイトーシス側へ放出されやすい、ということもあるかもしれません。
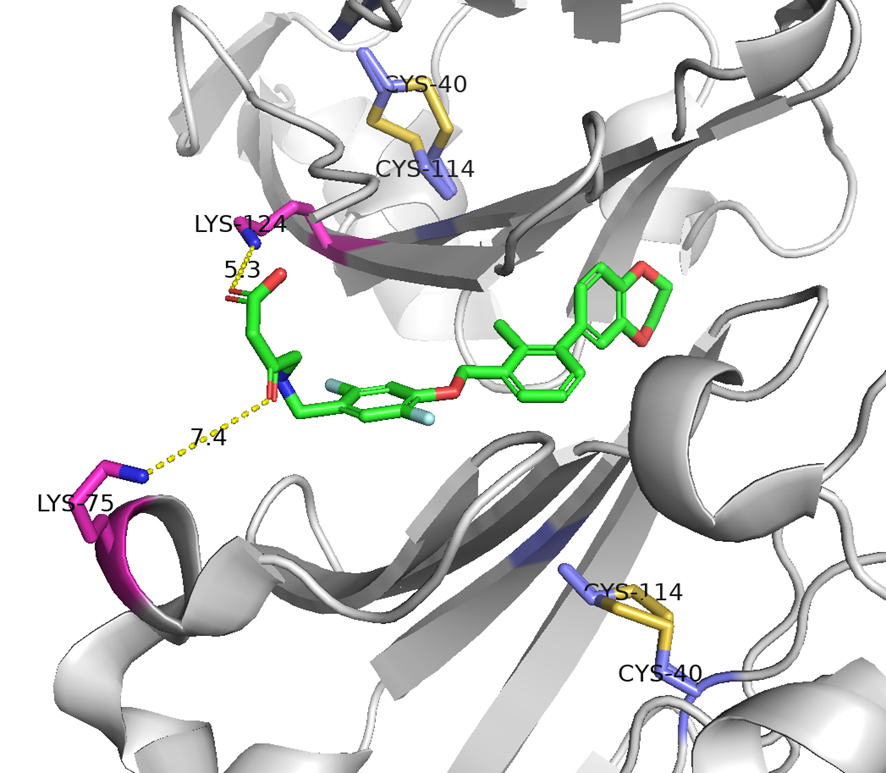
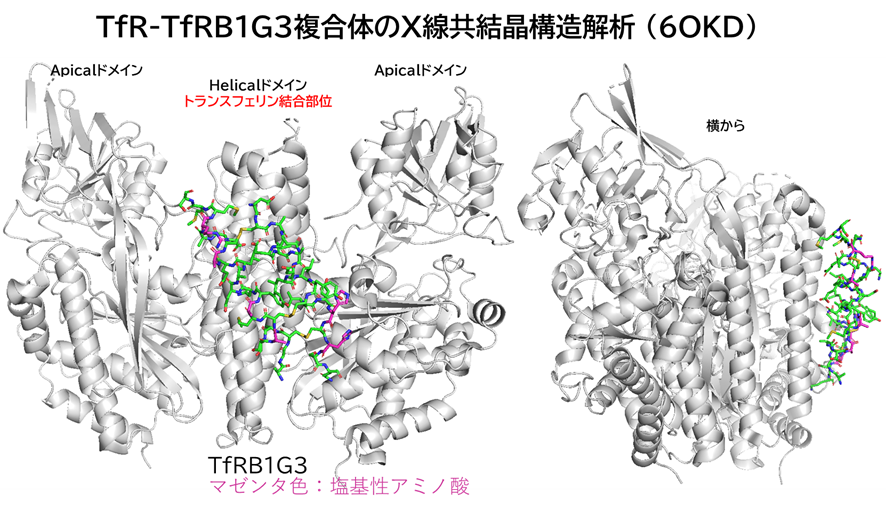
と言うのも、米フレッド・ハッチンソンがん研究センターのCrookらが報告したシステイン架橋ペプチドTfRB1G3は、アルギニンやリジン、ヒスチジンの塩基性アミノ酸を多く含んでおり(むしろアスパラギン酸やグルタミン酸の酸性アミノ酸の方が数自体は多かったが・・・)、TfRとの複合体のX線結晶構造解析によると、BCY17901とは異なり、トランスフェリンと同じ部位(ヘリカルドメイン)に結合しており、そしてBBB通過が示されたためです。
A TfR-Binding Cystine-Dense Peptide Promotes Blood–Brain Barrier Penetration of Bioactive Molecules
J Mol Biology, 2020, 432, 3989-4009.
https://doi.org/10.1016/j.jmb.2020.04.002.
と言うことで、hTfR1を標的としたBBB shuttleペプチドを手に入れるには、連続した塩基性アミノ酸を含むペプチドをデザインし、結合試験でトランスフェリンと競合するペプチドを釣ってこれば良いのかな?
【2026/06/26追記】
バナナさんのコメント
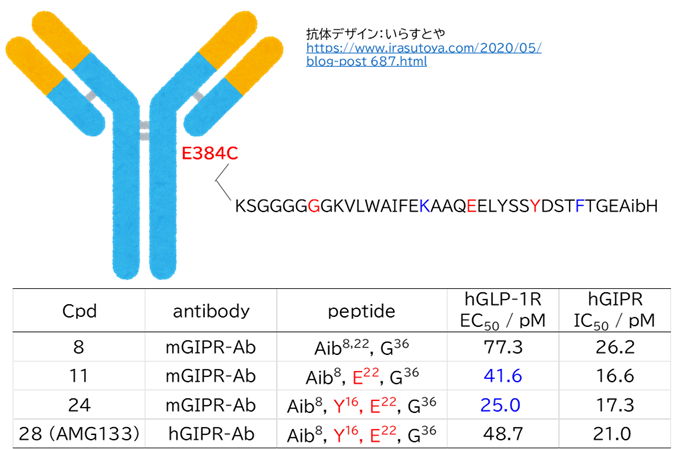
● 脳移行しやすい抗体のプロファイル
・ トランスフェリン非競合性エピトープ
・ TfR1に1価結合する
・ 低親和性(Koffが早い)
(ただし、中性と酸性で抗体の結合が大きく変わらない場合)
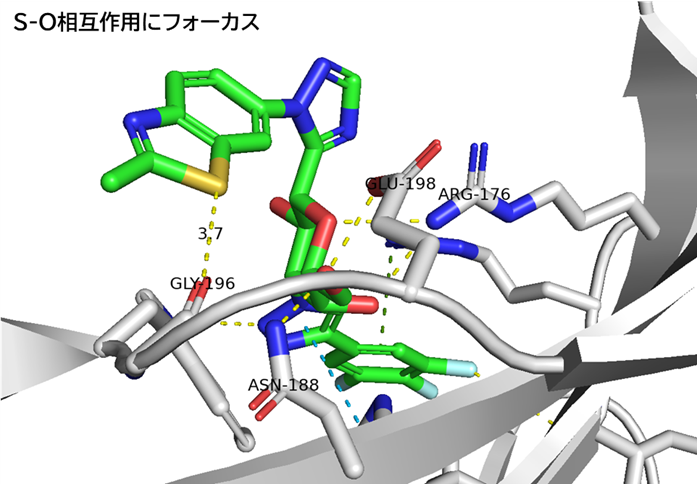
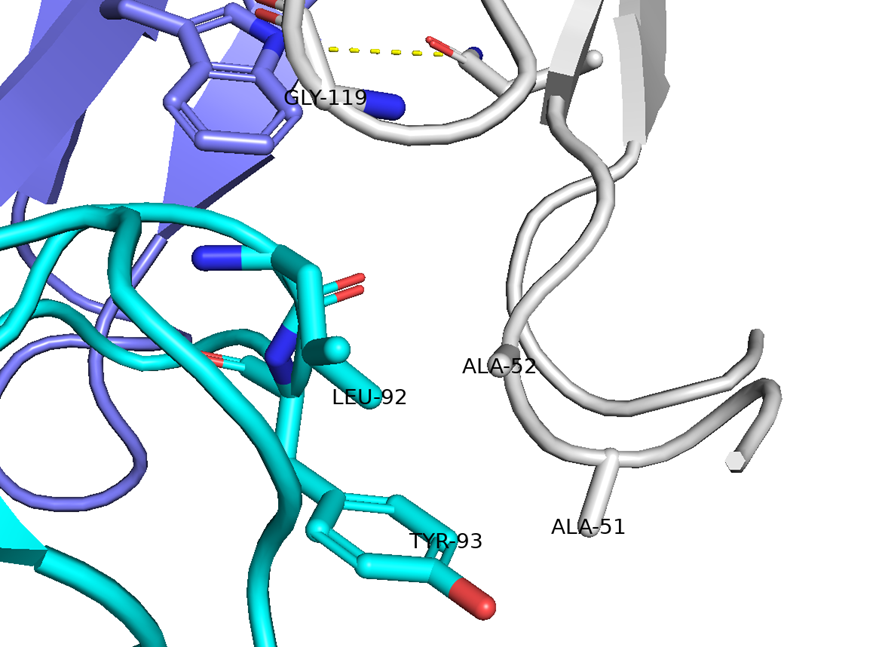
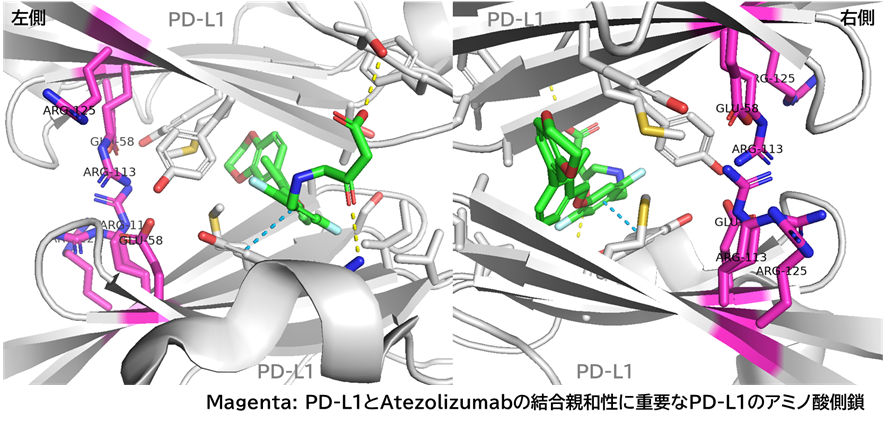
● No.894は高親和性でも、TfR1の非競合性エピトープ(apical domain)の露出しているTyr211&酸性アミノ酸(Asp204、Glu214、Asp302のどれか)と相互作用していて、pH依存性により脳移行が高いと想像
hTfR1-BCY15466複合体のX線共結晶構造解析 (9GH7)を元に、上記アミノ酸残基をピックアップして、hTfR No.894を重ねてみました。
(302 はAspでなくHisだったが、代わりにAsp284, Glu294 が周辺にあったので一緒にピックアップ)
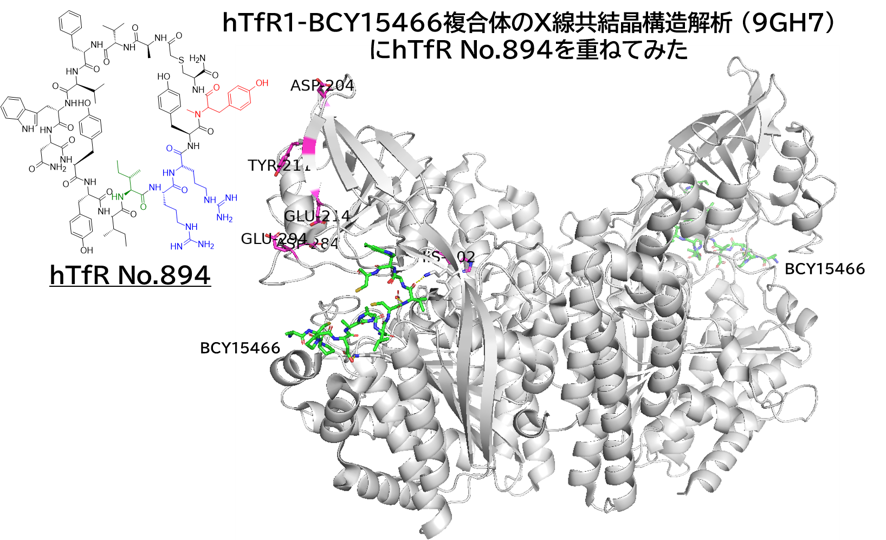
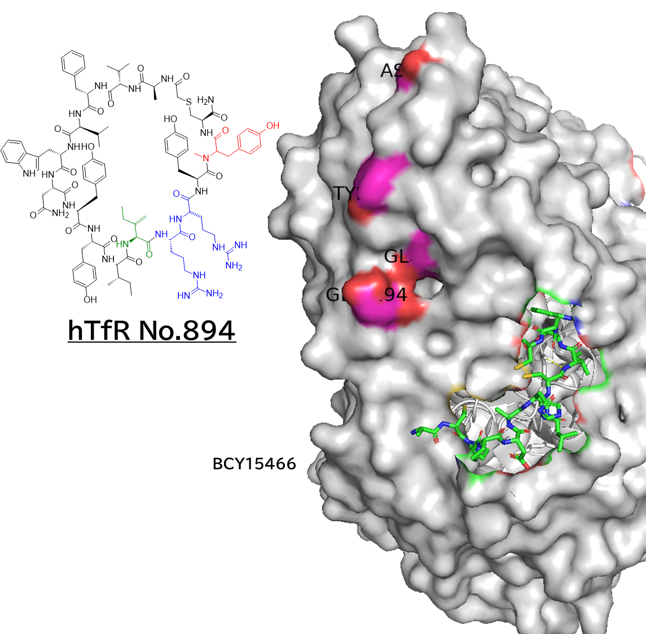
なんか相互作用しそうです。
興味深いのは、BCY15466の結合部位とは違うこと、そして、No.894は表面に露出した残基と相互作用しそうな一方、BCY17901はもっと下のポケットに深く入り込んでいることです。上でも書きましたが、疎水性+水素結合中心の induced fit で結合が安定し、解離が遅く、リサイクリング経路に留まりやすい解離しにくい(=筋移行しやすい)のが特徴かもしれません。
そうすると、結合部位および結合様式によって、つまりDDSペプチドのアミノ酸残基の組合せによって脳移行 or 筋移行を作り分けられるかもしれません。(と言っても、TfRはユビキタスに発現しているので、脳移行=筋肉や全身にもある程度行くとは思いますが)
そして脳移行TfRリガンドペプチドは、相互作用するアミノ酸残基が上側に集中しているので、シミュレーションでde novoデザインできるのでは? たとえば東京科学大の大上先生なら。
いやぁ、メドケムって本当にいいものですね。