やることは変わらない
環状ペプチドは、抗体のようにタンパク質間相互作用(結合部位が広くて平面)を選択的に狙いつつ、低分子のように細胞膜透過性があり経口投与可能で化学合成による低コスト化も期待できることから、長年注目のモダリティである。
今回は、一昨年巷を賑わせたMerckさんの環状ペプチドMK-0616創出の経緯である。大きな構造に圧倒されるが、ヒットペプチドからMK-0616取得までのメドケムアプローチは、低分子創薬と変わらない。
モダリティが変わろうが、創薬化学者がやることは変わらない。

Series of Novel and Highly Potent Cyclic Peptide PCSK9 Inhibitors Derived from an mRNA Display Screen and Optimized via Structure-Based Design
https://doi.org/10.1021/acs.jmedchem.0c01084
A Series of Novel, Highly Potent, and Orally Bioavailable Next-Generation Tricyclic Peptide PCSK9 Inhibitors
https://doi.org/10.1021/acs.jmedchem.1c01599
Orally Bioavailable Macrocyclic Peptide That Inhibits Binding of PCSK9 to the Low Density Lipoprotein Receptor
https://doi.org/10.1161/CIRCULATIONAHA.122.063372
PCSK9は、LDLコレステロール代謝に重要な役割を果たしている。
主に肝臓や小腸に発現しており、細胞表面にあるLDL受容体(LDLR)の細胞内分解を促進することで、LDLRによるLDLコレステロールのクリアランスを減少させる。
現在、PCSK9を標的とした脂質異常症の治療薬として、抗体医薬のEvolocumabや核酸医薬のinclisiranが承認されているが、PCSK9とLDLRとタンパク質間相互作用の結合サイトは大きく平面であるため、低分子バインダーは見つかっていない。一方で、承認薬の抗体や核酸は注射剤であるため、経口剤のアンメットメディカルニーズが存在する。
そこで、Merck社は経口吸収可能な環状ペプチドによりタンパク質間相互作用を阻害するアプローチを狙った。
余談だが、以前にPfizer社はPCSK9とLDLRのタンパク質間相互作用を直接狙うのではなく、その発現を抑制する低分子創薬(核酸医薬の低分子化)を報告している。
https://azarashi-panda.hatenablog.com/entry/2023/02/23/135326
【ハイライト】
1)X線複合体共結晶構造解析(と代謝部位同定)でメドケム方針を定める
2)不要な部分の除去、代謝部位周辺に置換基導入、環化して配座固定
3)極性基導入でOATP基質回避、製剤添加物で経口吸収化
【その1:X線複合体共結晶構造解析(と代謝部位同定)でメドケム方針を定める】
- Ra pharma社(UCB社が買収)とのコラボレーションにより環状ペプチド1を取得
- 2つのメドケムアプローチで化合物2を取得
- それぞれのアプローチで活性が大きく増加
- 配座安定化(エントロピー駆動型)に依る活性向上かな?
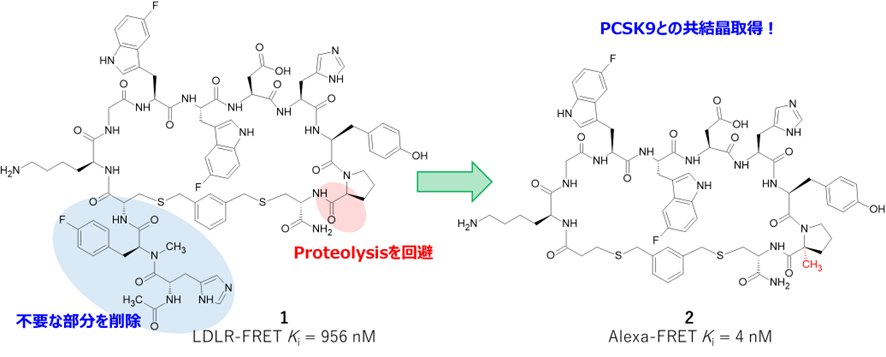
- 化合物2はPCSK9と共結晶を取得できたためX線解析を実施

【その2:不要な部分の除去、代謝部位周辺に置換基導入、環化して配座固定】
- 4つのメドケムアプローチで化合物17を取得
- 活性に不要な部分を除去して分子量を削減
- C末カルバモイル部分を除去
- 代謝部位周辺に置換基を導入して立体障害で代謝改善
- 環化して活性配座を固定
- オレフィンメタセシスで環化(transが活性体)
- ラットにアレルギー反応(マスト細胞脱顆粒)あり

- 化合物17は肝OATP基質(かつ阻害剤)のため高クリアランス
- ラットでOATPをKOするとCLが低減
- OATP基質性は脂溶性と相関
- 極性基を導入して回避を狙う(アニオントランスポーターなのでカルボン酸はダメ、マスト細胞脱顆粒リスクあるのでアミンの塩基性が高いとダメ)

【その3:極性基導入でOATP基質回避、製剤添加物で経口吸収化】
- 2つのメドケムアプローチで化合物44を取得
- 極性基を導入してOATP基質認識を回避
- 塩基性の低いアミンや3級アミンの導入が効果的
- リンカーをPEGにして更に極性化(溶解度向上に寄与)
- 環化して活性配座を固定
- アルキンとアジドでトリアゾールを形成するHuisgen反応で環化
- 極性基を導入してOATP基質認識を回避
- 化合物44はOATP基質認識を回避しつつ活性も大幅向上

- 環化を最適化してMK-0616を取得
- MK-0616は製剤添加物Sodium caprateで経口投与
- 第2b相試験で 高コレステロール血症患者のLDL-Cを有意に低下
https://www.msd.co.jp/news/chq-20230324-1/

今回、MerckのヒットペプチドからMK-0616取得までのメドケムアプローチを見ると、注目したい点は5つある。
①不要な部分を除去する
低分子創薬では、活性だけでなくLigand Efficiency(LE)を利用して(個人的には特にFBDDで使われる印象)、もし構造削減によって活性低減してもLEが維持されればヨシッとして合成展開することがある。ペプチドは元の分子量が大きいのでLEが使えるか不明だし、今回は構造削減によって活性が増強されたが、経口吸収化を狙うためには分子量を小さくしつつ水素結合ドナー/アクセプターを減らすアプローチは重要と思う。
②代謝部位周辺に置換基を導入して立体障害によって代謝回避を狙う
低分子創薬でも代謝部位またはその周辺に置換基を導入して、立体障害や電子吸引性を利用して代謝回避を狙うことはよくある。ペプチドでは、ペプチドの加水分解を回避するためにアミノ酸α位にメチル基を導入したり、2級アミノ酸のプロリンに変換したりするのが一般的のようだ。塩野義さんもやっていた。
https://doi.org/10.1021/acsmedchemlett.2c00310
とは言え、前提として代謝部位を同定する必要があるため、今まで紹介してきた塩野義さん中外さんJTさんと同様に、ヒット化合物の時点で代謝を評価することは重要だろう。ここを丁寧に取り組めるかが、今後の成功の可否を分けるポイントかもしれない。
③環化して配座固定する
これも低分子創薬でよくやる手法で、フレキシブルな構造をリジッドにして活性向上(や選択性改善、物性改善)を狙うが、今回は2回も環化することで活性を激増させた。ただし、何となく環化するのではなく標的タンパクとのX線複合体共結晶構造解析を元にデザインした方が良い。
④また、経口吸収する環状ペプチドと言えば、シクロスポリンや中外さん、ペプチドリームさん皆共通してN-メチルアミノ酸が含まれているが、今回のMK-0616には含まれていない(2級アミノ酸プロリンはあるけど)。これは何故だろうか?化合物2のX線解析から、PCSK9との相互作用に幾らか水素結合ドナーが必要っぽいが、それだけだろうか?分子内水素結合で配座の維持にも使われている?それともN-メチル化すると立体障害で配座が維持されない?中外さんのプラットフォームではアミノ酸 = 11個, CLogP ≧ 12.9, N-アルキル化 ≧ 6個が膜透過に重要とあったが、標的タンパクによってはもっと大きいサイズが必要だったり、N-メチル化できない場合もあったりするのかもしれない・・・と思った。
一方で、今回のMK-0616において5-フルオロトリプトファンのフルオロ基が活性に必須だったが、中外さんのペプチドライブラリーにもハロゲン化チロシンがあったので、ハロゲン(特にフッ素?)の入った非天然アミノ酸は重要かもしれない。あとプロリンも共通している。
⑤そして、論文を眺める限りでは経口吸収のためのアプローチは製剤添加物LabrasolまたはSodium caprateを加えるのみで、メドケム的に何を検討したのか分からなかった。おそらく2つの環化は効いているだろうけど。製剤添加物は、GLP-1受容体作動薬リベルサス(セマグルチド)もSodium Salcaprozate (SNAC)により経口化している。
以上、モダリティが変わっても創薬化学者がやることは基本的には変わらない・・・と言っても、こんな大きな分子を相手に同じことが出来るMerckさんはやっぱりスゴイ。Merckの化学は世界一チイイイイ!!